遺伝性ミオパチー
その他の遺伝性ミオパチー
2.筋原線維性ミオパチー
筋原線維の走行の乱れをふくむ変性像とそれにともなう他の細胞内小器官の変性像を主な形態変化とする一群の遺伝性ミオパチーを筋原線維性ミオパチー(myofibrillar myopathy)と呼んでいる(Nakano, 1996: De Bleecker, 1996; Selcen, 2004, 2011; Engel, 2004)。その一部では変異を示す遺伝子とそれがコードする蛋白が同定されているが、その多くはZ線を構成する蛋白またはそれに関連する蛋白である。一疾患群とはいえ、それぞれ異なる疾患であり、また未だに原因が明らかになっていない例も多く、今後の課題が多い。
臨床症状は緩徐進行性のミオパチーが主徴で、心筋症、不整脈、末梢神経障害などを伴う疾患がある。常染色体性優性遺伝をとる疾患が多いが、同劣性遺伝やX染色体性劣性遺伝をとるものもある。筋病理ではトリクローム染色で異常が検出されやすく、赤紫色の局面が筋形質にみられ、cytoplasmic body や nemaline rodsが時に観察される。大小の空胞もしばしば見る。また硝子様の局面が見られることもある。壊死線維が散見され、軽度の細胞浸潤を伴うことがある。組織化学的には筋線維内部の構築異常が顕著で、fiber splitting が見られる時がある。ミオパチーに加えて、神経原性変化が併存することがある。電子顕微鏡では筋原線維の走行異常、Z線のストリーミング、nemaline rod, 空胞変性、ミトコンドリアの形態異常、核の変性、線維性や顆粒状の封入体沈着などの多くは非特異的な所見が見られる。以下に現在原因が確認されている疾患を述べる。
(1)デスミン関連ミオパチー
デスミンは骨格筋、心筋、平滑筋に存在する横径約10 nmの中間径線維で、骨格筋ではZ線に近接して筋鞘下に豊富にあり、他の中間径線維とともに、筋原線維と筋鞘、核、ミトコンドリアなどとの構造的結合を保持している。
デスミノン関連ミオパチーは臨床的には小児から成人発症の慢性進行性筋萎縮を示し、萎縮の分布は多様で、遠位型、肢帯型、肩甲下腿、顔面などが報告されている。不整脈と心筋症を合併する頻度が高い。嚥下障害や呼吸不全を合併する例がある。多くは常染色体性優性遺伝をとるが、少数だが劣性遺伝もあり、また孤発例も多い。病理学的には筋原線維の配列異常にくわえて、デスミンの蓄積が筋形質内、とくに筋鞘下に見られることが多い(Goebel, 1995)。電顕でも筋原線維の走行の乱れ、cytoplasmic body などの筋原線維ミオパチーに共通してみられる変化がみられるが、加えて主に顆粒線維状の蓄積物が見られることが多い。
(2)αB-クリスタリノパチー
α-クリスタリンはheat shock protein の一つで、可溶性のオリゴマーを形成している。その機能は、変性した蛋白と結合して、異常な凝集体形成を防ぎ、細胞をストレスから守ることにあると言われている。αB-クリスタリンは骨格筋、心筋、眼の水晶体などに存在する。
αB-クリスタリノパチーは成人に発症し、骨格筋の萎縮を主徴とするが、白内障や心筋障害を伴うことが多い。常染色体性優性遺伝をとる。筋病理学的には筋原線維性ミオパチーに共通の所見に加えて、デスミノパチーで出現するものと類似した筋鞘下の顆粒線維様構造の蓄積を認めることが多い(Vicart, 1998)。
(3)ミオティリノパチー
ミオティリンはZ帯関連蛋白の一つで、Z帯を形成するα-アクチニンおよびフィラミンCに結合している。ミオティリン遺伝子の変異は肢帯型筋ジストロフィー1Aと分類されているように、40歳以降に発症する近位筋萎縮が普通であるが、後に下肢遠位部が加わることが多く、また構音障害や、心筋症、末梢神経障害も合併しうる。常染色体性優性遺伝を示す。筋病理では筋原線維ミオパチーの所見の中でもミオティリンばかりでなくデスミンやαB-クリスタリンなどの凝集体がみられ、電顕では空胞や線維性封入体が目立つ(Olive, 2005)。
(4)ザスポパチー
Z-band alternatively spliced PDZ motif-containing protein(ZASP)は骨格筋と心筋にあり、Z帯を形成するα-アクチニンと結合している。ZASP遺伝子の異常によるザスポパチ-は50歳以降に近位筋または遠位筋の筋力低下で発症することが多いが、中には原因不明の高CK血症として発見された例もある。心筋症や末梢神経障害を合併する例がある。常染色体性優性遺伝を示す。筋病理では筋原線維性ミオパチーの所見の中でも、小空胞が筋形質にみられることがある。Desmin, αB-crsystalin の他に、gelsolin, dystrophinaなどの凝集体がみられる。また電顕で thin filamentous inclusion を認める(Selcen,2005)。
(5)フィラミノパチー
フィラミンCは骨格筋と心筋に発現しており、アクチン線維のほかにミオティリンなどのZ帯関連蛋白、およびサルコグリカンと結合している。フィラミンC遺伝子異常によるフィラミノパチー家系はドイツや米国で発見されている。成人発症の主として近位筋の筋萎縮で、呼吸障害を来すことがある。また心筋症、末梢神経障害を伴う例がある。常染色体性優性遺伝を示す。筋病理では筋原線維の配列の乱れなどの筋原線維ミオパチーの所見があるが、縁取り空胞は少なく、 filamin C, desmin, myotilin, Xin, dystrophin, sarcoglycan の凝集体がみられる。電顕では tubulofilamentous inclusion が観察されるが、デスミノパチ-で観察されるものとの判別は困難と報告されている( Kley, 2007)。
(6)Bag3 異常症
Bcl-2 associated athanogene 3(BAG3)はCAI stressed-1 とも呼ばれ、HSP70 や抗アポト-シス蛋白 Bcl-2 その他と結合してシグナル伝達、抗アポトーシス、蛋白分解などに関与している。BAG3は骨格筋と心筋に多く発現している。BAG3異常症は小児で進行性筋萎縮を来たし、心筋症を合併し、呼吸不全にいたる重篤な症状を呈する。末梢神経障害を合併し、rigid spine を有する例がある。常染色体性優性遺伝を示す。筋病理では筋原繊維性ミオパチーに共通の所見に加えて、電顕でアポトーシスを起こした核が高頻度に観察される(Selcen, 2009, 2011)。
(7)FHL 1 異常症
Four-and-a-half-LIM domain 1 protein (FHL 1) はミオフィブリルと筋細胞膜に分布するタンパクで、その遺伝子 FHL 1 の変異は4つの異なる筋病態を引き起こすことが明らかになっている。それは
- 還元小体ミオパチー
- X染色体性抗重力筋萎縮型ミオパチー (XMPMA) (Windpassinger, 2008)
- X染色体性肩甲下腿症候群 ( Quinzii, 2008)
- エメリー・ドライフェス筋ジストロフィー(Schessl, 2011: Cowling,2011)
である。また rigid spine syndrome や心筋症も起こしうる (Schessl, 2011)。X染色体性優性遺伝をとる。重症度は乳幼児重症型から成人良性型まで様々である。筋病理学的にも多様だが、還元小体(Fig.38)の存在は重要で、その証明にはmenadione-linked α-glycerophosphate dehydrogenase (MAG) 活性の組織化学検査で、基質のα-glycerophosphate を使用せずに活性が見られる点が重要である(Fig.39)。しかし、エメリー・ドライフェス筋ジストロフィーとXMPMAの例では還元小体の存在は報じられていない (Cowling, 2011)。還元小体は核の近傍に存在することが多く、しばしばサイトプラスミック・ボディと共存する。電子顕微鏡では線維性構造の集合体で、その集合体は形も大きさも様々である(Fig.40).
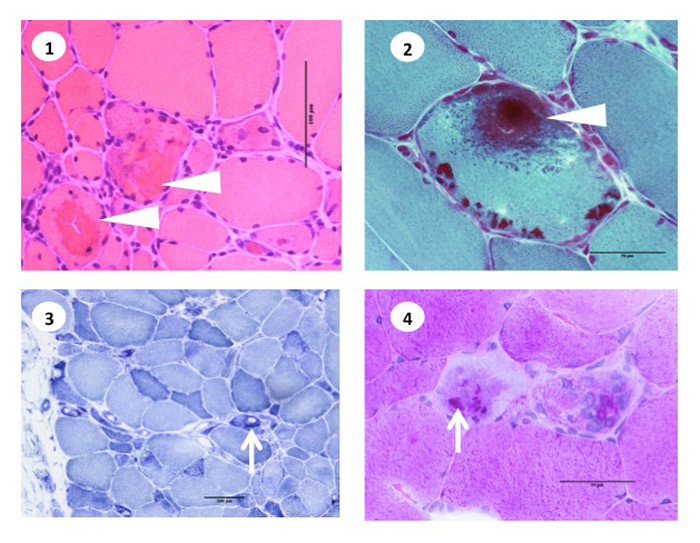
Fig.38
還元小体ミオパチー
- HE:還元小体は細胞質内で強い好酸性を示す(矢頭)
- トリクローム変法:還元小体は暗紫色に染まる(矢頭)。筋表面に近い、核近傍に好発する。
- NADH-TR 活性像:酸化酵素活性は低い(矢印)。
- PAS反応:グリコーゲン顆粒を伴っており、PAS反応で陽性をしめす。
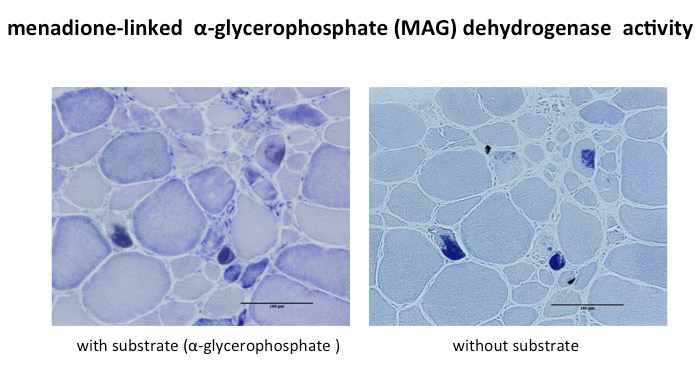
Fig.39
還元小体ミオパチーにおけるMAG活性像
メナジオンはSH基を含む構造物である還元小体に結合し還元される。テトラゾリウムは、還元されるとフォルムアザンを形成し発色する。このため、還元小体、テトラゾリウム、メナジオンが結合すると、基質として通常必要なグリセロール3リン酸なしで発色が見られる。

Fig.40
還元小体ミオパチ-の超微形態
①、②:還元小体の超微形態:線維の集合体が方向を定めず集積し(星印)さらに大きな集合体を形成し、その中にグリコーゲン顆粒の小集合体が散在している。大きな集合体の周囲には限界膜はみられない。
③:還元小体(星印)は核(N) 近傍によく見られ、しばしばサイトプラスミック・ボディ(CB) が共存する。周囲にZ線のストリーミング(矢印)や小さな縁取り空胞(矢頭)が見られることが多い。
(8)サイトプラスミック・ボディをともなうミオパチー
サイトプラスミック・ボディ(CB)はZ帯の構成物質に由来する構造で、筋細胞の非特異的な病理変化である。CSは炎症性筋症、各種の筋ジストロフィーおよび myofibrillar myopathy など多くのミオパチーで出現するだけではなく、神経原性筋萎縮でも見られることがある。Z帯と密接に関連した出現するので、CBの細胞内における分布はZ帯のそれに類似することがある。CSの出現を主要な病理変化とする遺伝性ミオパチーは従来から cytoplasmic body myopathy として報告され、その中には病初期から呼吸不全を来す例が多いことが注目されたが、その背景の病態は多様であると推定されていた。近年それに一致する臨床症状を呈しCS がみられる病態としてデスミン関連ミオパチー、ACTA1関連ミオパチー (Donervoort, 2017) および titin 遺伝子(TTN) の A-band ドメインの変異を伴う hereditary myopathy with early respiratory failure (HMERF) (Izumi, 2013) などが報告されている。これらはいずれも病理学的には myofibrillar myopathy の変化を示している。しかしHMERGの病態はなお多様と思われ、十分解き明かされてはいない。




