遺伝性ミオパチー
筋ジストロフィー
7.筋強直性ジストロフィー
筋収縮が遷延し、弛緩が遅延する現象で、筋電図で myotonic discharge が検出されるのが筋強直現象である(Fig.21)。筋強直現象(myotonia)を伴う筋ジストロフィーは大多数筋強直性ジストロフィー1型(MD1)であるが、欧州で報告された2型(MD2)もごく少数存在する。
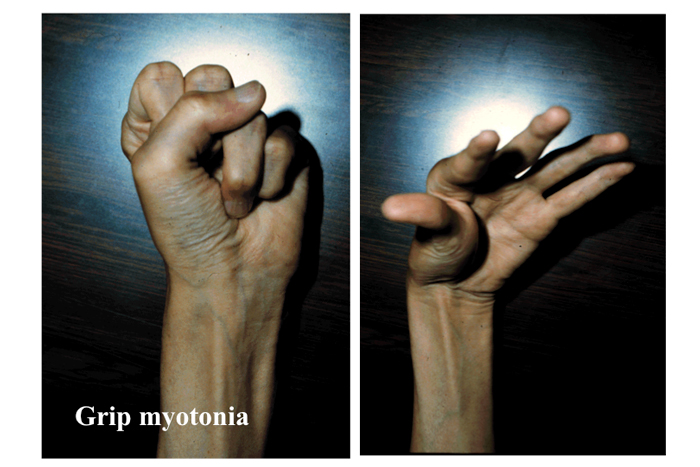
Fig.21
筋強直現症は収縮した筋の弛緩に時間がかかる現象で、強く握った指をほどくのが遅れる grip myotonia が見られる。
MD type 1
常染色体性優性遺伝を示す。発症年齢は胎児から高齢まで広い。しかし、出生前に発症する先天性筋強直性ジストロフィーについては、症状が出生後発症の例とは異なる点が多く、別に述べる必要がある。胎内ですでに胎動が乏しく、羊水過多を来しやすい。出生時にはフロッピーインファントの状態で、筋萎縮が目立ち、関節拘縮を伴う。しかし筋強直性現象は目立たない。テント状口などの顔貌の異常が見られ、知的および運動発達が遅滞する。母に何らかのMDの症状が見られ、母方祖父がMDに罹患していることが多い。この現象はCTG反復の伸張と性別の関連で説明できるとする仮説がある(Brunner, 1993)。
また発症年齢は同一家系では世代が下がるにつれて早くなる傾向(anticipation)がある。筋萎縮は顔面、頸部、四肢遠位筋に特に目立つ。心筋障害、心伝導異常、白内障、禿頭、および糖尿病など多臓器の異常が合併しやすい。また軽度の知能障害を合併することがある。血清CKは軽度から中等度に増加する。
染色体19qのdystrophia myotonica protein kinase 遺伝子(DMPK)の非翻訳領域にあるCTG反復配列の延長があり、これが転写されてできるpre-mRNAのCUG反復配列が、以下のような様々な蛋白生成にスプライシング異常を起こすと推定される。CLCN1(voltage sensitive chloride channel 1), RYR1(ryanodine receptor 1), MTMR1(myotubularin-related protein 1), INSR(insulin receptor), TNNT2(troponinT2), GDIN1(NMDA receptor subunit 1), APP(pre-amyloid beta A4), MAPT (microtubule-associated protein tau)
筋病理学的にはミオパチーの所見で、中心核が増加し、ring fiber などの筋線維内部構築異常が見られやすい。また type 1 fiber atrophy をみとめる(Fig.22)。
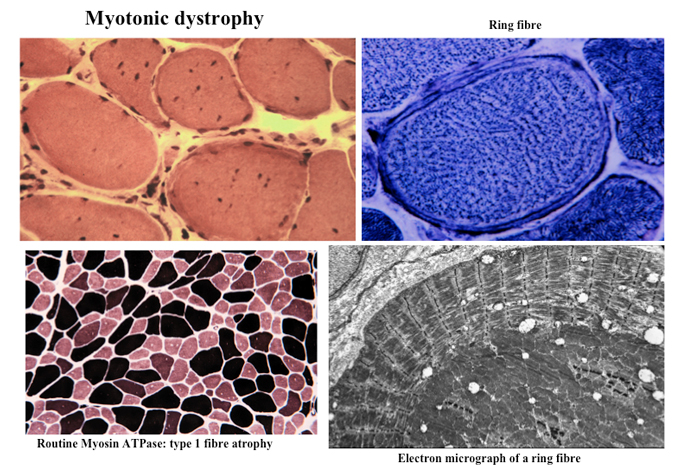
Fig.22
筋強直性ジストロフィーの筋はミオパチーの変化を示す。内在核が極端に増加し、リングファイバーとよぶはちまき様の構造が観察できる。これは筋原線維の一部が筋鞘下で走行方向の異常を来すためにおきる。また type 1 fibre 萎縮がみられる。
MD type 2 (proximal myotonic myopathy)
DM1と比較して発症年齢がやや遅く、先天性や若年性は見られない。Anticipation の傾向はあるものの、比較的めだたない。近位筋力低下優位で proximal myotonic myopathy(PROMM)とも呼ばれる。このためLGMDなどと誤診されやすい。筋痛が起きやすい。筋強直現象は一般に弱く、みとめないこともあるが、筋電図では記録できる(Ricker, 1994)。心筋障害、心伝導異常、白内障、禿頭、および糖尿病など多臓器の異常が合併しやすい(Ricker, 1995)。
染色体3q21の ZNF9(CNBP)遺伝子の非翻訳領域にあるCCTG反復配列の延長があり、これが転写されてできるmRNAのCCUG反復配列が様々なタンパク生成にスプライシング異常を起こすと推定される(Raheem, 2010) 。
筋病理所見はMD1と共通点が多いが、type 2 fiber atrophy をみとめることが多いと言われている。




