遺伝性ミオパチー
先天代謝異常によるミオパチー
2.脂質代謝異常
先天的脂質代謝異常症の種類は多いが、ミオパチーを起こす疾患は限られており、また頻度は一般に低い。ミオパチーに関連する脂質代謝異常症を表にまとめ、以下にその内重要なものについて述べる(Tab.3)。
Disorder of lipid metabolism of skeletal muscle
- Primary carnitine deficienty
- Carnitine palmitoyltransferase deficiency (CPT)
- 2.1. CPT 1 deficiency
- 2.2. CPT 2 deficiency
- Very long chain acyl-CoA dehydrogenese (VLCAD) deficiency
- Mitochondrial trifunctional protein deficiency
- 4.1. Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency
- 4.2. MTP deficiency (combined enzyme deficiency)
- Medium–chain acyl-CoA dehydrogenase (MCAD) deficiency
- Multiple acyl-CoA dehydrogenase deficiency
- 6.1. Riboflavin-insensitive multiple acyl-CoA dehydrogenase dificiency
- 6.2. Riboflavin-responsive multiple acyl-CoA dehydrogenase deficienty
- Other primary disorders of fatty acid oxidation
- 7.1. Carnitine/acylcarnitine transolocase deficiency
- 7.2. Short-chain acyl-CoA dehydrogenase deficiency
- 7.3. Medium-chain 3-ketoacyl-CoA thilase deficiency
- 7.4. Short-chain L-3-hydorxyacyl-CoA dehydrogenase deficiency
- 7.5. 2,4-Dienoyl-CoA redictase deficiency
- Other lipid storage myopathies
- 8.1. Muscle coenzyme Q10 deficiency
- 8.2. Chanarin-Dorfman disease or multisystem triglyceride storage disease
- 8.3. Neurtral lipid storage disease with myopathy
Tab.3
主な筋の脂肪蓄積症
(1)カルニチン関連蛋白異常症
カルニチンは長鎖脂肪酸と結合してそれを、細胞質からミトコンドリア外膜と内膜を通過し、ミトコンドリアのマトリックスに輸送する。この過程で必要なのはcarnitine transporter、carnitine palmitoyl transferase (CPT) I と CPT II、carnitine acyl carnitine translocase である。このカルニチンとその関連蛋白のうち、変異によるミオパチーを通常起こすのは全身性カルニチン欠乏症とCPT II 欠損症である。
i)全身カルニチン欠乏症
全身カルニチン欠乏症は細胞膜でのカルニチン輸送体である organic cation/carnitine transporter 2 のコード遺伝子 SLC22A2の変異によるもので、主に乳幼児に重篤な肝と心筋障害を起こすが、小児と成人発症型では比較的軽症で、心筋症に脂肪蓄積ミオパチーを伴う例がある。血清CKは小児発症で高いが、成人は症例では正常な例が多い。血中カルニチンの低値となる。
ii)カルニチンパルミトイルトランスフェラーゼII欠損症
CPT II は長鎖アシルカルニチンから長鎖アシルCoAを生成する。欠損症のうち新生児型と乳児型は重篤だが、筋型は主に小児から青年期に発症するが成人や中年発症の例もある。症状は筋痛や横紋筋融解の発作が繰り返されるのが普通で、間欠期には無症状で血清CK値も正常な事が多い。発作は運動、飢餓、寒冷、薬剤などに誘発されやすい。筋生検で見られる変化は普通正常か軽度のミオパチーのみである。血中アシルカルニチン分析がスクリーニングに行われるが、診断には組織の酵素活性測定が必要となる。
(2)β-酸化関連酵素異常
i)VLCAD欠損症と三頭酵素欠損症
アシルCoA のβ-酸化ではミトコンドリア内膜にある極長鎖アシルCoA脱水素酵素 (VLCAD) とそれに続くミトコンドリア三頭酵素 (TFP) が機能する。これらの先天的欠損症は筋症状を呈するが、TFP のドメインの一つである長鎖3ヒドロキシアシル-CoA 脱水素酵素 (LCHAD) のみの機能欠損例も海外で報告されているが、我が国では報告例はなく、TFP欠損例が多い。いずれにも心筋、骨格筋障害、脳症などを起こす重篤な新生児乳児型と、成人の骨格筋型があり、後者では横紋筋融解、筋痛、筋力低下などが見られる。新生児乳児型では低血糖がみられる。これら欠損症の新生児乳児および小児では肝腫大を伴う。TFP欠損症では末梢神経炎、網膜症を伴う例が報告されている。
ii)グルタル酸血症II型
電子伝達フラビン蛋白 (electron transfer flavoprotein : ETF ) はミトコンドリア内膜にあり、β-酸化経路、分枝鎖アミノ酸代謝経路などの複数の脱水素酵素から電子を受け取り、ETF脱水素酵素 (ETFDH) にそれを伝達することで、電子伝達系の中で重要な機能を果たしている。ETFまたは ETFDH の欠損による病態がグルタル酸血症II型、別名 multiple acyl-CoA dehydrogenase deficiency、である。上記の複数の脱水素酵素の活性が低下し、臨床症状として重症の新生児型では筋緊張低下、呼吸障害、低血糖がみられ、一方、遅発型では運動後などに遅発型の筋痛、横紋筋融解などが起きる。筋病理ではtype 1、2線維ともに脂肪蓄積が見られるが、他に特異的変化に欠ける。リボフラビン、L-カルニチン、グリシンが治療に有効なことがある。
(3)中性脂肪分解関連酵素異常
i)リピン欠損症
リピンは細胞質にあってはホスファチジン酸経路のトリアシルグリセロール(旧名トリグリセリド)合成を促進する一方、核内因子としてその分解を促進する。リピンをコードする遺伝子の一つであるLIPIN1変異があると反復性の横紋筋融解が起きるが、それを誘発するのは運動ではなく感染や発熱が多い。間欠期の筋病理では正常か、脂肪蓄積を見ることがある。
ii)中性脂肪蓄積病
トリアシルグリセロールの分解に関連する酵素欠損により起きるまれな病態が Neutral lipid storage disease (NLD)(Fig. 35)であるが、現在同定されているのは魚鱗癬を合併する NLSDI(Chanarin-Dorfman syndrome)とミオパチーと心筋症を起こしやすい NSLDM である(Schweiger, 2009)。
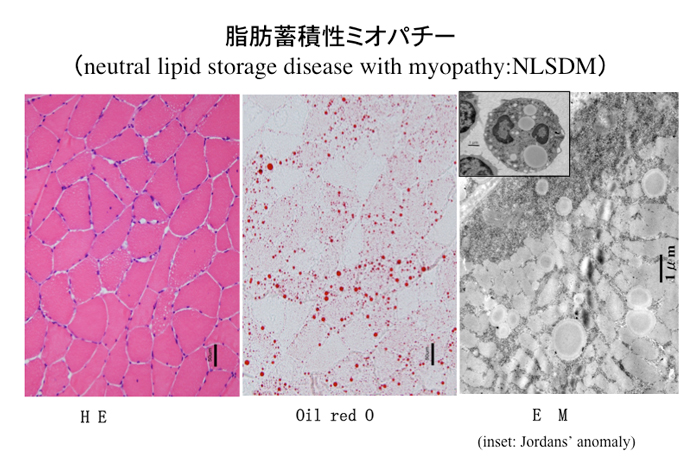
Fig.35
トリグリセリド蓄積症の一型であるneutral lipid storage diseaseの生検像。脂肪染色(oil red O)と電顕では大小多数の脂肪滴がある。また多角白血球の細胞質に脂肪滴がみられる Jordans anomaly が観察できる(EM像左上の挿入写真)。本例は遺伝子変異が同定されていないが、NSLDM と推定される。
前者ではcomparative gene idenfication-58(CGI-58)をコードする遺伝子α/β-hydrolase domain-containing protein 5(ABHD5)に異常があり(Lefevre, 2001)、また後者では adipose triglyceride lipase(ATGL)をコードするpatatin-like phospholipase domain containing 2(PNPLA2)に変異が見られる(Fischer, 2007)。CGI-58はATGLを活性化する作用があると考えられている。いずれの病態も常染色体性劣性遺伝を示し、上記の症状に加えて、難聴、脂肪肝をともなうことがあり、また多核白血球に脂肪滴が観察される(Jordans’ anomaly)。




