遺伝性ミオパチー
先天性ミオパチー
5. 管状凝集体ミオパチー myopathy with tubular aggregate (MTA)
管状凝集体(tubular aggregate)は凍結切片のHE、トリクローム染色で細胞質に青黒く染まる局面をつくり、NADH-TR活性像では強い活性を示すが、succinic dehydrogenase(SDH)の活性は示さない(Fig.28)。
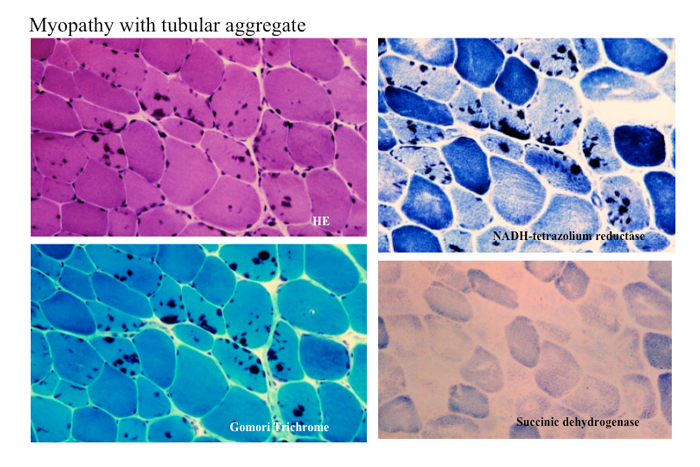
Fig.28
姉妹で筋無力症状と近位型筋萎縮を示した例。筋病理では主に type 2 fibre にHE、トリクローム、NADH-TRで黒く染まる一方、SDHでは染まらない構造がある。これは蓄積しているのが管状構造凝集体 tubular aggregate であることを示唆している。
これはこの構造がミトコンドリアではなく小胞体に由来することを示唆している。実際小胞体に分布するリアノジン受容体抗体に親和性を示す(Fig.29)。
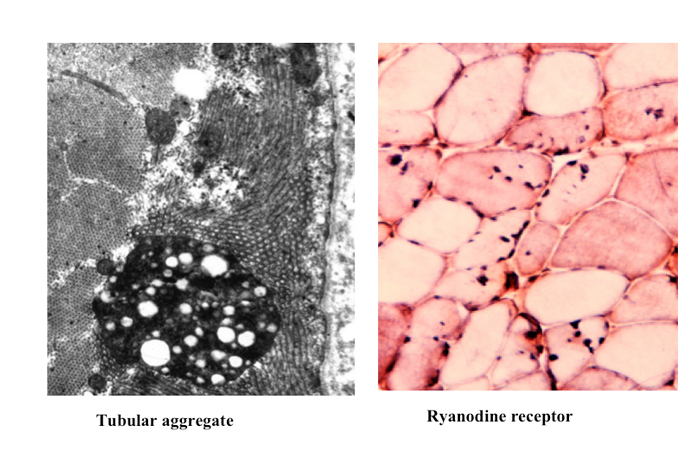
Fig.29
電顕では管状構造物の凝集体 tubular aggregateがあり、抗ryanodine receptor抗体で染色された。筋無力症状をともなう病型では小胞体のカルシウムセンサーであるstromal interaction molecule 1 (STIM1)のほか数種類の遺伝子変異が報告されている。
管状凝集体の出現を主な病理所見とする先天性ミオパチーを myopathy with tubular aggregate または tubular aggregate myopathy と呼んでいる。しかし、管状凝集体は低カリウム性周期性四肢麻痺や先天性筋無力症候群(Furui, 1997)などの各種のミオパチーで出現が報告されていて、疾患特異性はない。
先天性管状凝集体ミオパチーには(1)筋力低下のみまたはそれに筋痛を伴う病型と、(2)筋力低下と筋無力症状を伴う病型が知られている。
(1)には筋小胞体にあるカルシウムセンサーであるstromal interaction molecule 1(STIM1)(Bohm, 2013)と、その刺激を受けて細胞膜上で細胞外カルシウムを取り込む(store-operated calcium entry)カルシウムチャネルの構成蛋白ORAI1の遺伝子変異(Endo, 2015)などによって発生する。
(2)は筋小胞体で蛋白の糖鎖修飾に必要な酵素GFPT1(Senderek, 2011)とDPAGT1(Belaya, 2012)の遺伝子異常により起きることが報告されている。




