遺伝性ミオパチー
筋ジストロフィー
5.肢帯型筋ジストロフィー
肢帯型筋ジストロフィー(limb-girdle muscular dystrophy: LGMD)は、少なくとも25をこえる異なる疾患を類似する症状に基づいて寄せ集めた疾患群である。その概念はWaltonとNattrass(1954)を中心に1950年代に形成されたが、その後、変遷を遂げている。初期の概念は ” 小児期(1歳台の後半)から成人まで、ときに中年で、下肢帯または上肢帯の筋力低下で発症するもので、大多数は常染色体性劣性遺伝を示す。通常は緩徐進行性で、ときに高度の障害や死をもたらす ” 疾患であった。しかし、症例の蓄積とともに概念の境界は拡大し、1995年のENMC LGMD consortium では ” 四肢近位筋を主とする筋力低下をきたし、CK が高く、筋病理がジストロフィー変化を示す病態 ” と再定義されるに至った(Bushby, 1995; Straub, 2018)。
(1)病態
代表的な病型の遺伝子座、関連する蛋白質、臨床的特徴などを表(Tab.1)にまとめた。
常染色体優性遺伝
| 遺伝子座 | 変異蛋白 | 主な臨床症状など | 同一遺伝子による病態 | |
|---|---|---|---|---|
| LGMD1A | 5q31 | Myotilin | 近位筋、鼻声 | myofibrillary myopathy |
| LGMD1B | 1q11-q21 | Lamin A/C | 近位筋、心筋、不整脈 | AD-EDMD, CMT2B, lipodystrophy, cardiomyopathy, progelia |
| LGMD1C | 3q25 | Caveolin 3 | 近位筋、下腿肥大 | rippling muscle disease, distal myopathy |
| LGMD1D | 7q | Desmin | 下肢帯、上肢帯 | |
| LGMD1E | 6q23 | DNAJB6 | 近位筋、心筋 | |
| LGMD1F | 7q32.1-q32.2 | Transportin | スペイン人症例、近位筋 | |
| LGMD1G | 4q21 | HNRPDL | ブラジル人症例、指拘縮 | |
| LGMD1H | 3q25.1-p23 | 不明 | イタリア人症例 近位筋、下肢、下腿肥大 |
常染色体劣性遺伝
| 遺伝子座 | 変異蛋白 | 主な臨床症状など | 同一遺伝子による病態 | |
|---|---|---|---|---|
| LGMD2A | 15q15 | Calpine-3 | 近位筋、翼状肩甲 | |
| LGMD2B | 2p13 | Dysferlin | 近位筋、遠位筋 | Miyoshi myopathy, distal myopathy |
| LGMD2C | 13q12 | γ-sarcoglycan | 近位筋、下腿肥大 | |
| LGMD2D | 17q12-21 | α-sarcoglycan (adhalin) | SCARMD | |
| LGMD2E | 4q12 | β-sarcoglycan | ||
| LGMD2F | 5q33-34 | δ-sarcoglycan | ||
| LGMD2G | 17q11-12 | Telethonin | 下肢近位・遠位筋、上肢近位筋 | |
| LGMD2H | 9q31-34 | TRIM32 | 米在住ハッター症例 | |
| LGMD2I | 19q13.3 | Fukutin-related protein | 上肢近位筋、下肢 | |
| LGMD2J | 2q24.3 | Titin | フィンランド人症例、前脛骨筋 | |
| LGMD2K | 9q34.1 | POMT | トルコ人症例、知能障害、幼児発症 | |
| LGMD2L | 11p13-p12 | Anoctamin 5 | フランス系カナダ人、近位筋 | |
| LGMD2M | 9q31 | Fukutin | 日本人、幼児発症、知能障害なし | 福山型先天性筋ジストロフィー(FCMD) , cardiomyopathy |
| LGMD2N | 14q24 | POMT2 | 筋緊張低下、運動発達遅延、翼状肩甲 | α-dystroglyconopathywith mental retardation (CMDB2) |
| LGMD2O | 1p32 | POMGnT1 | 小児発症、近位筋、翼状肩甲、足関節拘縮 | |
| LGMD2P | 3p21 | α-dystrooglycan (DAG1) | 小児発症、近位筋、知能障害、足関節拘縮 | |
| LGMD2Q | 8q24 | Plectin | 運動発達遅延 | CMD with familial junctional epidermolysisbullosa |
| LGMD2R | 2q35 | Desmin | 顔面、近位筋、呼吸筋、心筋 | |
| LGMD2S | 4q35.1 | TRAPPC 11 | 顔面、近位筋、知能発達遅延 |
Tab.1
主な肢帯型筋ジストロフィー
(2)筋病理
肢帯型筋ジストロフィーにおける筋病変は病型により違いがあるが、全般的な共通点もある。本節でまず共通的な所見を述べて、次項で各病型の特徴について述べる。
共通して見られるのは慢性に結果するミオパチーの所見である(Fig.17)。
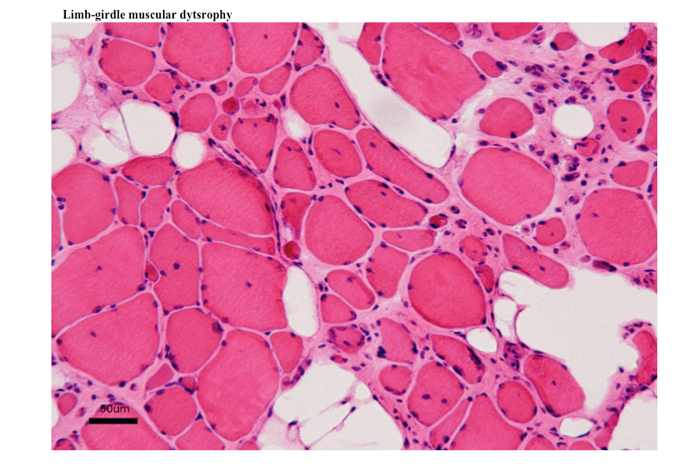
Fig.17
肢帯型の1例の筋生検. 筋線維の横断面は円形化し、横径に大小不同が見られる。極度に萎縮した線維
から正常径、さらに肥大線維まで偏りなく存在する。内在核が高頻度の見られ、また間質の開大と線維化をみとめ
る。白く抜けた空間が目立つのは脂肪沈着の部位。いずれも慢性のミオパチーを示す所見である。
筋線維横径の著しい大小不同は、萎縮線維とともに肥大線維が多いことによる。肥大線維は fiber splitting を起こすことがある。中心核は増加し、しばしば中心核を一端に持つ、あるいは中心核を通過する fiber splitting を見ることがある。また、程度は様々だが間質には線維化と脂肪織の沈着が見られるのが普通である。
(3)症状と筋病理各論
我が国で頻度の多い病型について臨床的特徴を要約するとともに各病型における筋病理と免疫組織化学の所見について述べる。
i)LGMD1A
ミオティリンはZ帯に存在し、他のZ帯形成蛋白と結合しながらアクチン線維の構造を維持するために重要な役割を果たしている。染色体5q31に位置するミオティリン遺伝子の変異により LGMD1A とmyofibrillar myopathy が起きると考えられている(Salmikangas, 1999) (Selcen D, 2004)。その臨床症状は40歳台以降に起こる遠位筋または近位筋の筋力低下または運動に誘発される筋痛で発症し、緩徐に進行して全身に広がり、一部に呼吸筋を障害する。遠位型の一部に声帯と咽頭筋を障害するものがある(Salmikangas, 1999)。心筋障害や末梢神経障害をともなう(Olive, 2005)。
筋病理学的には rimmed vacuole を含む空胞形成をともなう myofibrillar myopathy の所見で、超微形態はZ line streaming などの局所的筋原線維変性と空胞形成が中心である(Carisson, 2007)。免疫組織では局所的にmyotilin は量的に増加している。しかしこのmyotilin の増加はネマリン・ミオパチーや central core disease でも報告されている (Schroder, 2003)。
ii)LBMD1B
ラミンA/Cは核膜を構成する蛋白の一つで、核骨格蛋白としての働きばかりでなく、蛋白合成などに関与すると考えられている。遺伝子は1q11-q23にあり、その変異に伴って見られる病態は多系統に及び、近位型筋萎縮を主徴とするLGMD1B (Kitaguchi,2001)、常染色体優性遺伝型エメリー・ドライフェス症候群 (Bonne, 1999)、拡張型心筋症、心伝導ブロック (Sinagra, 2001)、家族性局所性リポジストロフィー (Cao, 2000)、下顎先端異形成症 (Novelli, 2002)、ハッチンソン・ギルフォード早老症 (Kirschner, 2005)、軸索型シャルコー・マリー・トゥース病2C型 (Goizet, 2004)、その他である。
LGMD1Bの筋病変は比較的穏やかなミオパチー変化にとどまる例が多い。電顕では核膜の変化が見られる (Matsubara, 2004)。しかし、免疫組織学的にはラミンA/Cの変化は通常の方法では異常を検知することは困難である。
iii)LGMD1C
カベオリン-3(caveolin-3: CAV3) は筋細胞膜にある50-100 nm の陥凹部を構成する蛋白で、細胞膜を通過する物質の輸送とシグナル伝達に役割を果たすと考えられている。またジスフェルリンをはじめとする細胞膜蛋白と密接に連関する。ヒトのCAV3遺伝子は3p25にあり、その変異はLGMD1C (Minetti, 1998)の他に本態性高CK血症とrippling muscle disease (Betz, 2001)を起こす。
LGMD1Cの臨床症状は小児期に起きる近位筋の筋痙攣や筋萎縮のことが多い。遠位型筋萎縮を示すこともある。Rippling muscle disease では筋線維の被刺激性がまし、軽い叩打などの刺激で局所の筋線維が収縮し、わずかな隆起が周囲にされさざ波のようにその筋の辺縁に向かって広がる現象がみられる。
筋病理学的には非特異的なミオパチーの所見があるが、免疫組織学的には筋線維表面にCAV3が検出できない。しかしCAV3の量の減少はLBMD2Bでも二次的に観察される。
LGMD1C の臨床症状にリポジストロフィーを伴う例で、caveolin-associated protein polymerase I and transcript release factor (PTRF 別名 cavin) の遺伝子変異が報告されている(Hayashi. 2009).
iv)LGMD1D,E,F,G
LGMD D は7q36にあるDNAJ subfamily B member6 (DNAJB6) 遺伝子の変異を伴う(Sarparanta, 2012)。DNAJB6は熱ショック蛋白40ファミリーの属するJ蛋白の一種でZ帯に分布し、分子シャペロンとして蛋白の結合や折りたたみなどの過程に関与すると考えられている。欧州神経筋カンファレンス(ENMC) は2018年のLGMD ワークショップで本病態を” LGMD D1 DNAJB6-related myofibrillary myopathy (LGMD D1 DNAJB6関連筋原線維性ミオパチー)“と呼ぶことを提唱した (Straub, 2018)。なお、この新分類が定着する時期が来たら、本稿でもそれに従い改訂する予定である。筋病理学的にはサルコプラスミック・マスやリングファイバーが出やすく、分葉状筋線維も出現する。
LGMD1Eでは成人発症で緩徐進行性筋力低下が下肢からはじまり、拡張性心筋症と心伝導障害を伴う。時には遠位型筋力低下を示す。筋病理学的には自己融解空胞と蛋白凝集体を伴う myofibrillary myopathy を呈すると考えられている。
LGMD1F では7q32.1-7q32.2 にあるトランスポルチン (transportin:TNPO) 3 遺伝子に変異がある。この蛋白は核膜に存在しタンパク質の核内への移行に関与する (Melia, 2013; Torella, 2013)。臨床的には発症は乳児から中年までと巾があり、普通は下肢の筋力低下から発症し、その分布は近位筋と遠位筋のいずれの優位もある。少数だが翼状肩甲、眼瞼下垂、呼吸不全などを伴う例がある。筋病理学的にはミオパチー性の筋変性像にくわえて、筋核内に横径10-20 nmの線維性封入体がみられ、同時に rimmed vacuole が観察された。
LGMD1G は南米で少数例が報告されたのみのまれな病態で4q21に位置する遺伝子の変異によるとされている (Starling, 2004)。個々に位置するHNRPDL遺伝子のコードする蛋白はリボ核蛋白ファミリーに属しmRNA合成や代謝に関与する。臨床的には成人発症の下肢から始まる近位筋萎縮が主症状で、白内障と2型糖尿病を合併しやすい。筋病理的には軽度のジストロフィー変化、すなわち筋壊死や間質線維化をともなうミオパチーで、HNRPDLは筋核周囲に染色されるが、患者では正常より強く染まりやすいと報告されている(Vieira, 2014)。
LGMD1H では中年発症の緩徐進行性近位筋力低下が主症状である。関連遺伝子は 3p23-p25.1 にあると報告されている (Bisceglia, 2010).
v)LGMD2A
LGMD2Aで欠損するカルパイン3(CAPN3) はカルパイン・ファミリーに属するカルシウム依存性の細胞内蛋白分解酵素で15q15.1にある遺伝子CAPN3でコードされている。CAPN3は筋細胞の維持に不可欠と考えられているが、その詳細は不明で、普段は不活性な細胞質内の質が、必要時にCAPN3によって活性化され、筋線維を維持するとの仮説がある (Beckmann, 2008)。
発症年齢の幅は広いが、多くは10から15歳で、腰帯筋とくに内転筋群と大殿筋の筋力低下で発症する。大腿外転筋群(小殿筋、中殿筋、梨状筋など)が比較的保たれることが多い。走ることと階段の昇降が不自由となり、動揺性歩行が見られるようになる。翼状肩甲がおこりうる。下肢近位筋の筋力低下が進行するが、下腿筋の肥大がある例は少数で、あっても程度は軽い。一方、足関節の拘縮が見られることは多い。血清CK活性は高いが、LGMD2Bほど高くはない。重症度にはおおきな個人差があるが、多くは30歳前後に車いすを必要とする。心筋障害はまれで、知能障害はない (Fardeau, 1996)。
筋病理では壊死線維と再生線維があり、筋内鞘に線維化をともなう間質の開大がおきる。Type 1 fiber predominance が見られるのが普通で、筋線維の多くはlobulated fiber である。免疫組織化学では dystrophin, utrophin, sarcoglycan は正常に筋線維表面に局在する。Calpine 3 は他の筋ジストロフィーの原因蛋白のように筋膜や核膜に分布する構造蛋白ではなく、筋形質に分布する不安定な酵素蛋白であるため、抗体による免疫染色を使っての診断は確立しておらず、実施しても結果の解釈に困難がある。診断は主に immunoblotting によって行われている。
vi)LGMD2B
ジスフェルリン異常症の症状は多様で、肢帯型の例もあるが、遠位型の分布を示す三好型筋ジストロフィーの病型をとるものもある。十歳台で非常に高い血清CK活性を示し、本態性高CK血症として検査を受ける例が多く、その後年齢と共に次第に下肢筋力低下が明かになる経過をたどる。肢帯型の病型を示す患者でも遠位筋に萎縮が見られる例が多く、下腿筋の仮性肥大は見られない。20歳台から少しずつ筋力低下が目立ち始める例が多いが、進行速度と重症度には個人差が大きい。まれに高齢発症例が報告されている (Klinge, 2008)。
筋病理ではミオパチーの変化が見られるが、高CK血症のみの段階では筋変性は比較的軽度である。しかし、しばしば局所的に炎症像が散見される例があり(Fig.18)、炎症性筋症との鑑別が問題となる。筋病理像から肢帯型と三好型を区別することはできない
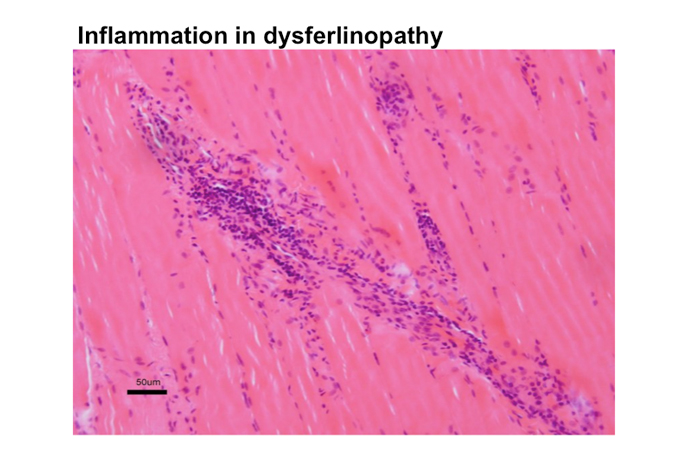
Fig.18
LBMD2Bの一例:ミオパチーの変化に加えて軽度の細胞浸潤が見られる。
免疫組織化学では、抗 dysferlin抗体に対する筋表面の染色では染まらない例が多い(Fig.19)。
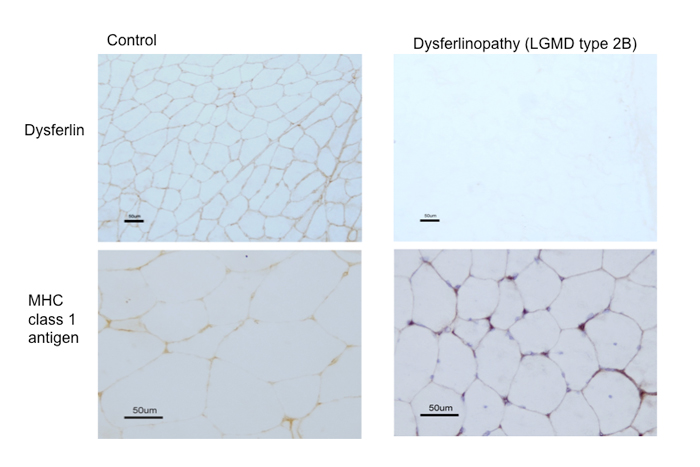
Fig.19
肢帯型の病型特定には免疫組織化学と遺伝子解析で試みられる。このLGMD2Bの例ではジスフェルリンの欠損が推定され、遺伝子検査でもtype2Bと確認された。この病型は、下段の様にMHC class I 抗原が軽度に異常発現することがある。
筋ジストロフィーをふくむ他の筋疾患でも二次的にdysferlinの染色性の低下がおきることがあるので、判断には慎重さが求められる。一般に免疫染色はスクリーニング検査と考え、確定診断のためには遺伝子解析が必要である。
遺伝子異常に関してはLGMD2Bと三好型遠位型筋ジストロフィーの異常に差はなく、同じ遺伝子異常でもいずれも起こりうるとされている(Illarioshkin, 2000)。遺伝子解析でcompound heterozygote が確認できる例が66%、一つの病的アリルが確認できた例が22%との報告がフランスからある (Krahn, 2009)。保因者の病態については十分確認されていないが、 筋のdysferlin タンパクが半減していたとの報告がある(Fanin, 2006)。
vii)LGMD2C, D, E, F
サルコグリカン(sarcoglycan)複合体は主にα,β,γ,δの4つのサルコグリカンからなる複合体で、ジストロフィン結合糖タンパク複合体 (dystrophin-associated glycoprotein complex) の一部を形成し、筋膜下の細胞骨格蛋白と細胞表面を結ぶ重要な役割をになっている。サルコグリカン異常によるLGMDは他の病型(2A、2Bなど)と異なり、臨床症状で下腿筋の仮性肥大が85%と高頻度にみられ(Eymard, 1997)、時に巨舌をみるなど、ジストロフィン異常症と類似した点がある。
αサルコグリカノパチー(Adhalinopathy) は本邦では筋ジストロフィーの約2%に存在すると報告されており(Hayashi, 1995)、 severe childhood autosomal recessive muscular dystrophy (SCARMD)とも呼ばれ、DMDやBMDと臨床症候の類似性が高い。
生検筋の変化はLGMD2A, 2B と比較して強いことが多く、壊死・再生が活発であるのに対して、lobulated fiber の頻度は比較的低く、type 1 fiber predominance が比較的弱い。免疫組織化学的検査では、抗アダリン(α- sarcoglycan)抗体に対して、反応性が失われたものが53%あった一方、残る47%では反応の減弱が見られた(Eymard , 1997)。このようにどのサブユニットに変異があっても、他のサブユニットの染色性も低下することが多いが、当該のサブユニットの低下が最も激しい。
viii)LGMD2G, H,I,J,K,L,M
LGMD2Gはテレトニン(teletonin)遺伝子の変異で発生する(Vanizof, 2002)。TeletoninはZ線に存在する19-KDの蛋白でtitin をZ帯に固定する役割があるといわれている。LBMD2Gは常染色体劣性遺伝LGMDの約2.7%を占め、症状は近位筋または遠位筋萎縮を呈し、翼状肩甲と下腿の筋肥大を伴いやすいとともに下垂足を呈することもある。 LGMD2G患者骨格筋では免疫組織化学的には抗体に全く反応しないか、きわめて弱い反応をする(Francis, 2014)。
LGMD2HはE3-ubiquitin ligase である tripartite motif containing 32 protein(TRIM32)geneのpoint mutation によって発生する(Frosk, 2002)。1976年にカナダ在住のフッター派の人々に見られたLGMDの特徴が報告され、1998年にはその遺伝子座が9q31-33にあることが、さらにそれがTRIM32遺伝子の変異によることが明らかになった。変異のみられるNHLドメインはユビキチン化の標的蛋白との結合部位と推定されている。TRIM32はLGMD2Hの他にアメリカのフッター派住民(Jerusalem, 1973)と南ドイツ住民で報告されていたsarcotubular myopathyの原因遺伝子であることも明らかになった。LGMD2Hの主症状は緩徐進行性の近位筋萎縮で、しばしば顔面筋を障害し、翼状肩甲や腓腹筋の肥大と足関節拘縮を伴うことがある(Shieh, 2011)。顔面肩甲上腕型の分布を示す例も報告されている。血清CK活性の増加は比較的軽度である。筋電図では筋原性と神経原性変化が混在し、筋病理学的にはミオパチーであるが、vacuolar myopathy を呈し、fiber type grouping を伴うことがある。一部の保因者の筋にはvacuolar myopathy が見られといわれている。
LGMD2J はtitin 遺伝子(TTN)の変異でおきる稀な病態である。TitinはZ帯を構成する分子の一つで、LGMD2Jの病理像には myofibrillar myopathy と共通している点が多い。
LGMD2I、LGMD2KおよびLGMD2Mはそれぞれ fukutin related protein(FKRP), protein-O-mannosyl transferase 1(POMT1)および fukutin遺伝子の変異によるもので、いずれも筋表面の細胞骨格蛋白であるα-dystroglycan (ADG)の糖鎖形成(glycosylation)に必要と考えられる蛋白である。それらの変異のため細胞表面の基底膜の形成が障害され、筋細胞の脆弱性がおきると考えられており、この一群の疾患はα-dystroglycanopathy と総称されている(Muntoni, 2008)。
LGMD2I はアジアでは比較的まれであるが、欧米では頻度の高いLGMDである。フランスの報告では(Bourteel, 2009)幼児期から20歳台までに、平均年齢9歳で運動時筋痛や易疲労性で発症し、その後下肢近位筋の筋力低下を主徴とし、上肢近位筋も障害され、一部で翼状肩甲を示した。進行性で平均20歳ごろ車椅子を要した。その後大多数の例で呼吸不全が30歳頃から出現し、発症平均16年で一部に夜間NIPPVを使用するに至った。少数例で心筋障害や不整脈が見られた。筋病理ではジストロフィーの変化で、免疫組織学的にはα-dystroglycanの染色性が低下ないし斑状(patchy)であった。
LGMD2JはZ線に存在する蛋白質である titin の遺伝子変異による稀な疾患で、このため筋構造蛋白の異常をきたすmyofibrillar myopathy の一つである。Myofibrillar myopathy について別項で述べる。
LGMD2Mの原因であるfukutin遺伝子異常およびLGMD2Kの原因であるPOMT1遺伝子異常については先天性筋ジストロフィーの項で述べる。なおLGMD2Kについては同じ遺伝子異常による先天性筋ジストロフィーである Walker-Warburg syndrome よりも臨床症状の比較的軽い、知能障害を伴ったトルコ人の例が報告されている(Dincer, 2003; Balci, 2005)。Fukutin遺伝子の異常は、最初重篤な運動障害と知的障害をともなう福山型先天性筋ジストロフィー(後述)で見いだされた。しかし、その後次第に多様性が明らかとなった。すなわち知的障害や大脳形成異常をともなわず、骨格筋症状が比較的軽症で肢帯型筋ジストロフィーの病像を呈するか、あるいはほとんど目立たない状態で、拡張性心筋症が主症状、ときには唯一の症状である例が見いだされるようになった。この状態がLGMD2Mと呼ばれている (Murakami, 2006; Godfrey, 2006)。
LGMD2P とLGMD2Q について少数の報告があるが、未だ十分な知見がないので、本稿では記載しない。




