遺伝性ミオパチー
筋ジストロフィー
8.先天性筋ジストロフィー
元来、出生時に症状が明瞭な筋ジストロフィーを先天性筋ジストロフィーと呼んできた。しかし、分子遺伝学的な知見が深まるとともに、先天性筋ジストロフィーと、生後一定期間をおいて発症する肢帯型筋ジストロフィーや先天性ミオパチーの境界は不明瞭になってきた。現在なおこのカテゴリーを維持する意味は、頻度は少ないものの、筋症状が重篤であるだけでなく、しばしば中枢神経系や目の異常を伴うこの疾患群が、臨床的にきわめて重要であることにある。以下に米国神経学アカデミー(AAN)の分類に基づいて述べる。
(1)コラーゲン VI ミオパチー
筋線維間に存在する膠原線維は筋の機能発現のためには不可欠な存在と考えられている。その主な構成成分であるコラーゲンVI遺伝子(COL6A1, COL6A2, COL6A3) の変異により起きるCMDが重篤なウルリッチ型CMDと比較的軽症のベスレム・ミオパチー(Bethlem myopathy)である。もともと別疾患と考えられていたが、両者の中間的な症状を示す例も存在し、同一遺伝子COL6Aの異常による病態であることが明らかになった。ウルリッチ型CMDがARとADの遺伝形式をとるのに対し、ベスレム型の多くはADをとる。
ウルリッチ型CMDでは生下時より下肢帯などの近位関節に拘縮がある一方、手指などには関節の過伸展がみられる。成長とともに筋力低下、脊柱側弯などが目立つようになり、呼吸不全となる例がある。また皮膚に瘢痕ができやすい。ベスレム・ミオパチーでは生下時に手指の拘縮が見られることがある。10歳頃から足や肘関節などの拘縮と、近位筋を主とする筋力低下が出現しする。筋力低下は30歳以降に進行が目立つようになり、60歳頃には歩行困難となる例が多い。症状は個人差が大きく、一部の患者では軽度の呼吸不全が生ずる。その後肢帯型筋ジストロフィーの病像を呈する例や軽症の常染色体性劣性遺伝の筋硬化症を呈する例も報告されている。
筋病理ではウルリッチと Bethlem 両病型ともに非特異的なミオパチーの所見を示すが、 ウルリッチ型では病初期には type 1 fiber atrophy と predominance を示し、fiber type disproportion の状態を呈しやすいという報告がある(Schessl, 2008)。ウルリッチ型の免疫組織化学では抗collagen VI抗体に対する反応が消失するAR例が報告された(Higuchi, 2001)一方、ADの例では間質にcollagen VIは検出されるものの、二重染色法で筋線維基底膜とcollagen VIを染色すると、正常対照のような筋線維基底膜とcollagen VIの共存が見られない例がある(Pan, 2003)。 COL6A 遺伝子内の変異部位などの差によるもと説明されている。しかし、ベスレム・ミオパチーでは、この二重染色法でも判断が困難な例がある。
以上のように全貌は明らかではないが、筋線維間のマトリックスにあって微少線維ネットワーク(microfibrillar network)の形成に重要な役割を果たし、筋線維の基底膜とも密接に関連する collagen VI の変異が、筋線維の変性のみならず腱、軟骨、皮膚および椎間板などの異常を来している可能性が考えられている(Lamande, 2018)。
(2)メロシン欠損症とインテグリンα7欠損症
欧米に多いCMDで、臨床症状は生下時からの筋緊張低下、筋萎縮、関節拘縮、呼吸不全が見られるが、心筋障害や知能障害は原則としてみられない。
筋病理的にはジストロフィーに共通な変性・壊死と再生を伴うミオパチーの変化があり、間質の線維化が高度である。Type 1 fiber atrophy が見られやすい。免疫組織学的にはlaminin-α2(merosin) が筋細胞表面に欠損している(Tome,1994)。ただし、次項でのべるFCMDでも二次的な欠損が部分的に見られることがある。本邦では、α-dystroglycan とともに筋細胞のlaminin 受容体である integrin α7の遺伝子異常においてCMDが発見されている(Hayashi, 1998)。これについても免疫組織学的な検査が可能である(Pegoraro, 2002)。
(3)α-ジストログリカノパチー
α-ジストログリカンは筋線維の表面構造の形成と維持に重要な役割を果たしている。このタンパク質は翻訳後にグリコシル化やマンノシル化などの翻訳後修飾を受けるが、この修飾にかかわる酵素の遺伝子異常により発生する種々の先天性筋ジストロフィーが知られている。
i)福山型先天性筋ジストロフィー
日本で診療の対象となる先天性筋ジストロフィーの大多数は福山型筋ジストロフィー(FCMD)である。生下時に筋萎縮、関節拘縮がみられ、中枢神経の発達異常による知能障害がみられ、生後さらに呼吸障害などの合併症が進行する。眼球運動障害、皮質盲、網膜剥離など、視力に関する異常を呈する例が約半数ある。しかし、遺伝子診断の発達により、比較的症状が軽く進行が遅い例もあることが次第に認識されるようになり、このような例の診断も重要となっている。常染色体性劣勢遺伝形式をとり、原因遺伝子は9q31にあるフクチン(Fukutin)遺伝子である。その3’領域のnoncoding region に3kb のretrotransposon の挿入がみられる例が多く、その homozygote の例が多数を占めるが、その他の点変異や欠失などの変異もあり、一部の患者はこれらの変異と挿入変異との複合ヘテロ接合体(compound heterozygote)である(Toda, 1999)。
フクチン蛋白の機能と局在は十分解明されていないが、細胞外に分泌され(Kobayashi,2001)、筋細胞膜と基底膜の結合を安定化する機能を持ち、他のα-ジストログリカノパチーの原因蛋白と同様、α-ジストログリカンのグリコシレーションに関与すると推定されている(Yamamoto, 2010)。
筋病理ではジストロフィーの所見である。免疫組織学的には基底膜の成分である laminin、なかでもlamininα2 の分布が不規則で断続的になり、全体量が減少する(Fig.23)。電顕では基底膜の連続性が失われ、欠損部位が広く観察できる。欠損部位の筋細胞膜が不明瞭になるなど変性がみられる (Matsubara, 1999)。
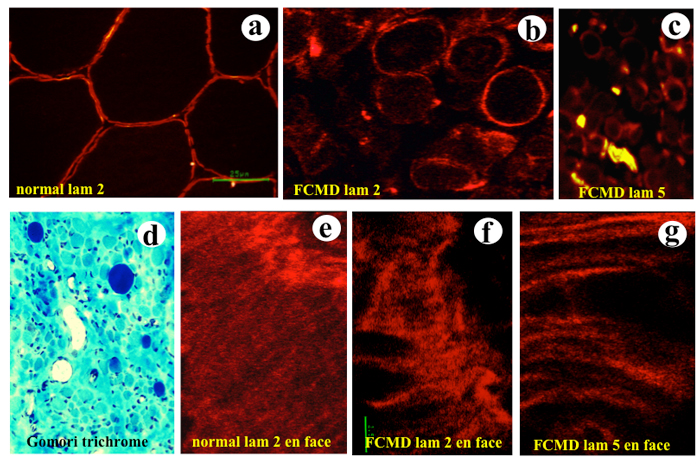
Fig.23
筋細胞基底膜に結合するラミニンを染色すると正常では細胞全周に染まりますが、福山型では欠損部位が目立
ちます。
a:正常対照 ラミニン2、b:FCMD ラミニン2、c:FCMD ラミニン 5、d:FCMD Gomori trichrome、E:正常対照 ラミニン2 en face 像、 f:FCMD ラミニン2 en face 像、 g:FCMD ラミニン5 en face 像
ii)Muscle eye brain disease(MEB)
MEBはじめにフィンランドから報告されたCMDだが、糖鎖形成にかかわる glycosyltransferase である protein O-mannose β-1,2-N-acetylglucoseaminyltransferase 1(POMGnT1)遺伝子に種々の異常が見いだされた後、その臨床症状が多様であることと、日本を含む世界各地に患者が存在することが明らかになった(Taniguchi, 2003)。大多数の例は生下時から筋緊張が低下し、生後も臥床状態で成長しても立つことは困難である。また視力障害が高度で知能発達も障害され、てんかん発作を有する例もある。目は高度の近視、視神経低形成や網膜の剥離や色素沈着がみられる。脳には小脳と大脳に形成異常がある。POMGnT1活性の低下が報告されている。
筋病理はα-ジストログリカノパチーに共通点が多く、免疫組織化学的なラミニンやαジストログリカンの局在については、抗体の認識部位による差があるなど鑑別には困難があるため、診断は遺伝子解析が必要である。
iii)Walker-Warburg syndrome(WWS)
脳回欠損症、滑沢脳症、高度の水頭症、クモ膜嚢胞などの中枢神経形成異常にともなう重症のCMDとして報告されていたWWSと福山型CMDやMEBの異同について検討されてきた結果、WWSではαジストログリカンの糖鎖形成に作動するprotein O-mannosyltransferase 1(POMT1)遺伝子に異常を持つ家系が見いだされた。臨床症候についてはα-ジストログリカノパチー間に類似性があるが、典型例とされる報告例に関する限り、中枢神経の異常は他疾患とは異なっていて、高度の水頭症やクモ膜嚢胞など特徴的な変化が含まれている。筋病理についてはMEBと同様と考えられており、診断には遺伝子検査が必要である。
iV)その他
α-ジストログリカノパチーには上記の他に FKRP, LARGE、POMT2 遺伝子その他の変異例も発見され、その多様性がさらに明らかになっている。
Fukutin related protein(FKRP)遺伝子変異は肢帯型筋ジストロフィー、LGMD2I(既述)の原因となるが、CMD1Cの原因でもある。FKRP はジストログリカンの糖鎖形成に関与すると推定され、ゴルジ装置に分布する。FKRP遺伝子異常はときにMEBやWWSの表現型をとるなど、単一の遺伝子変異としてはきわめて多彩な症状を現す特徴がある。FKRP遺伝子異常によるCMDとLGMDでは骨格筋症状とともに心筋や中枢神経の障害が起きうる。LGMD2Iの筋病理ではジストロフィーの所見で特異性に乏しい。免疫組織学的にはlaminin-α2 の染色性は不定だがα-dystroglycanの染色性の低下する例が多いと報告されている。
ヒトのLARGE遺伝子は第22染色体にあり、ヒトでは5番目に大きな遺伝子である。Large蛋白にはα-glycosyltransferaseの機能があると推定されている。マウスに自然発生した中枢神経異常を伴う筋ジストロフィーの一つにLARGE遺伝子に欠失があるものがあることから、神経障害を伴うヒトCMD遺伝子をスクリーニングした結果1例が発見された(Longman, 2003)。
(4)Rigid spine with muscular dystrophy と Multiminicore myopathy
脊椎強直(rigid spine)は特異的な症状ではないが、それを伴うCMD は従来よりDubowitz らにより一疾患として提唱されてきた。それをうらづけたのは selenoprotein 遺伝子に確認された変異(Moghadaszadeh, 1998,1999)である。Selenoprotein は主に小児で骨格筋などの小胞体に分布する蛋白で、筋の成長発達に関与すると推定されている。生下時より筋の発達が悪く、rigid spine とともに脊柱側弯、亀背などがあり、重症例では呼吸不全が合併することがある。症例間で重症度に大きな差があり、他の遺伝的素因の関与も推定されている(Moghadaszadeh, 1998)。筋病理学的には非特異的なジストロフィーの変化と報告されているが、同じ遺伝子に変異を持つ例の中に筋線維の内部構築以上の一つである multi-minicore やMallory bodyを呈するものが報告されSEPN related myopathyという疾患群名も提唱されている(Ferreiro, 2004)。
Multicore disease は A.G.Engel ら(1971)が小児で筋線維内に微少なミトコンドリア酵素活性を欠くコアが多発する良性の病態を報告したことに端を発するが、その後追加の報告として複数の遺伝子異常を伴うミオパチーで報告された。その中にはSEPN1, RYR1, MYH7などの遺伝子異常が含まれている(Cullup, 2012)。従って、独立疾患ではなくmulti-minicore myopathies と呼ぶのが適切と思われる。




