Non-hereditary Myopathies
Idiopathic inflammatory myopathies
9. Other inflammatory myopathies
(1) Granulomatous myopathies including sarcoid myopathy
Sarcoid myopathy or sarcoid myositis refers to sarcoidosis of the skeletal muscle, which presents with a histopathological presentation of non-caseating granulomas with giant cells in the muscle tissue. A few patients might show granulomatous lesions in the muscle without features of sarcoidosis in other organs—commonly known as granulomatous myopathy or granulomatous myositis.
Granulomatous and sarcoid myopathies are often indistinguishable histopathologically. Generally, patients can be categorized into two groups as those with prominent inflammatory cell infiltration with degeneration and regeneration in several muscle fibers and those with relatively sparse inflammation and mostly unaffected muscle fibers except fibers compressed by the granulomas (Fig. 53).
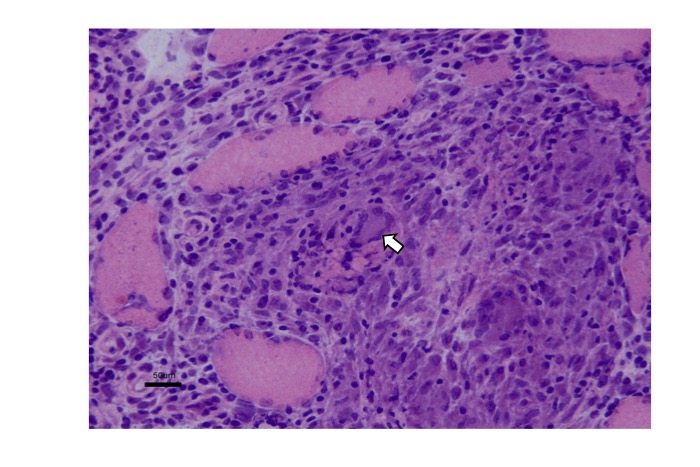
Fig.53
Granulomatous myopathy, including sarcoid myopathy, show non-caseating granuloma with multinucleated giant cells of Langhans type (arrow) and epithelioid cells. Degeneration of muscle fibers of varying degrees can be seen.
The former condition needs to be differentiated from Toxoplasma-induced changes and other conditions associated with eosinophilic infiltration.
(2) Focal myositis
Focal myositis is associated with acute or subacute localized swelling typically in the neck, which often spreads to the face and arms, usually associated with local pain and fever (Cain, 1998). It occurs both in adults and children and may occur in a variety of conditions. Myopathological changes include degeneration and regeneration of muscle fibers, infiltration of macrophages and lymphocytes and fibrosis of the interstitial tissues. Corticosteroids are usually effective. A small number of patients with eosinophilic infiltration and patients with neurogenic changes have also been reported (Auerbach, 2009). Focal myositis has been implicated in patients who subsequently developed PM (Heffner, 1981), as well as in those showing head drop secondary to age-related neck muscle weakness (Kastrup, 2008).
(3) Eosinophilic myositis
Eosinophilic myositis includes a wide spectrum of conditions causing myopathy associated with eosinophilic infiltration (Kaufman, 1993). They include parasitic infection, toxic myositis, vasculitis (Selva-O’Callaghan, 2014) and fasciitis. Idiopathic eosinophilic myositis refers to eosinophilic infiltration and inflammation of skeletal muscles in the absence of an identifiable cause. It may or may not be associated with eosinophilia or hypereosinophilc syndrome. A small number of patients showing focal myositis have been reported (Kobayashi, 2001). A study has reported a subacute presentation in a patient showing myalgia, fever, peripheral neuropathy, and myocardial involvement (Layzer, 1977).
(4) Myositis associated with chronic graft-versus-host disease
Reportedly, the 5-year incidence of chronic graft-versus-host disease (GVHD) was as low as 0.05% in skeletal muscle and fascia after the introduction of allogeneic peripheral blood stem-cell transplantation (Allo PBSCT) (Oda, 2009). However, with increasing numbers of patients undergoing such treatment, it is important to be aware of this condition. Myositis and fasciitis can occur simultaneously; therefore, yellow discoloration of fascia and joint contractures need to be carefully examined. Myositis shows considerable similarity to that observed in patients with PM except that forearms are commonly involved. Histopathological examination of muscle specimens also shows similarity between this condition and PM with infiltration of CD8+ cells (Parker, 1996; Stevens, 2003) (Fig. 54).
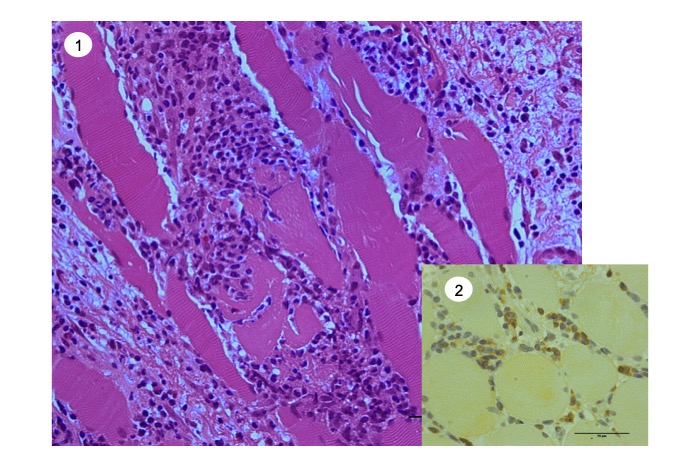
Fig.54
Chronic graft-vesus-host disease of skeletal muscle. (1) Inflammatory cell infiltration and degeneration of muscle fibers. (2) CD8+ cytotoxic T cells are seen.
Myositis occurs commonly in association with GVHD in other organs, although it can occur independently. Muscle biopsy is a useful diagnostic aid, and corticosteroids or immunosuppressants are used for treatment.
(5) Immune checkpoint inhibitor-induced inflammatory myopathy
Programmed cell death 1 (PD-1) inhibitors have revolutionized cancer therapy owing to their outstanding effectiveness. However, among adverse effects caused by these agents, inflammatory myopathy is considered one of the most disabling. It usually manifests approximately a month (mean) after initiation of PD-1 inhibitor therapy. Patients present with muscle weakness and elevated serum CK levels. It is occasionally associated with myasthenia gravis (Seki, 2019).
Histopathological findings include scattered foci of degenerated muscle fibers and inflammatory cell infiltrates. The infiltrating cells include CD8+ cells, CD4+ cells, macrophages, and B cells. Aberrant expression of MHC class I antigen is observed on the surface of muscle fibers (Fig. 55).
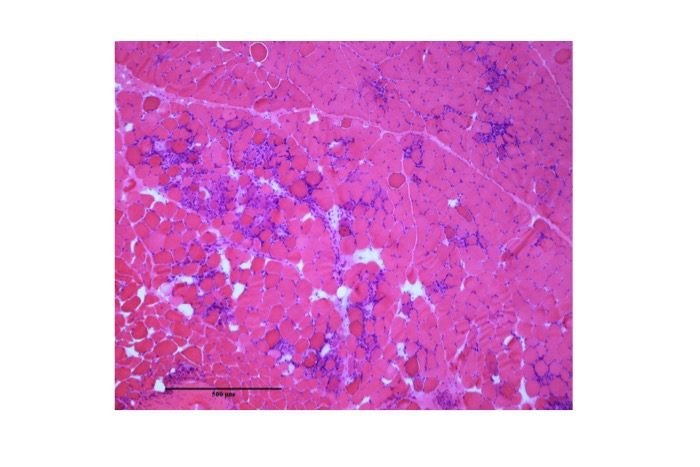
Fig.55
Immune checkpoint inhibitor-induced inflammatory myopathy. Foci of inflammation and degeneration of muscle fibers are seen.
(6) Vasculitis affecting muscles
Vasculitis is a clinicopathological process characterized by inflammation of and injury to blood vessels (Harrison’s Principles of Internal Medicine, 20/E). It can be categorized as primary and secondary vasculitis. These changes occur in the skeletal muscle and the histopathological changes in muscle are similar to all forms of vasculitis regardless of its cause. Therefore, it is possible to diagnose vasculitis histopathologically, but it is often difficult to confirm its cause exclusively based on morphological features. We discuss two relatively common types of primary systemic vasculitis.
i) Polyarteritis nodosa and microscopic polyangitis
Small-to-medium sized arteries are predominantly affected in polyarteritis nodosa (PN) and microscopic polyangiitis (MPA), which are often indistinguishable histopathologically. Capillaries and venules in the vicinity can also be affected by MPA. The edematous vessel wall narrows the lumen, and inflammatory cell infiltrates are identified in the vessel wall with fibrinoid degeneration. Structural destruction of the vessel wall can be confirmed by histopathological examination of the elastic lamina using Elastica van Gieson stain in paraffin-embedded sections. Polymorphonuclear neutrophils may be identified during the acute phase of PN; however, these are later replaced by mononuclear lymphocytes. Various degrees of perivascular inflammatory cell infiltration is identified. Generally, muscle fibers are affected to a limited degree; however, necrotic fibers can be observed in severe cases. Type 2 fiber atrophy is common in many types of systemic arteritis. Neurogenic changes are common owing to concomitant peripheral nerve involvement. Perifascicular atrophy is rare. Immunohistochemical examination reveals aberrant expression of the MHC class I antigen on the surface of the muscle fibers in the vicinity of the affected blood vessels or inflammatory foci.
ii) Eosinophilic granulomatosis with polyangiitis (EGPA) (Churg-Strauss syndrome)
Churg-Strauss syndrome is characterized by asthma, peripheral blood and tissue eosinophilia, extravascular granuloma formation, and multi-organ vasculitis. Histopathological findings include vasculitis involving small-to-medium sized arteries, veins and venules associated with eosinophilic infiltration in and around the vessel wall and formation of granulomas with epithelioid and giant cells around the blood vessels. Fibrinoid necrosis of the vessels is common. Changes in muscle fibers resemble those observed in other types of vasculitis.




