Hereditary myopathies
Metabolic myopathies
3. Autophagic vacuolar myopathies
Protein degradation is an indispensable function of eukaryotic cells. Autophagy constitutes an intracellular pathway for protein degradation and is initiated with the formation of autophagic vacuoles by molecules that are coded by a group of genes referred to as ATG genes. Autophagic vacuolar myopathies are congenital myopathies characterized by excessive formation of autophagic vacuoles.
Autophagic vacuoles first surround the target proteins or other debris with double membranes, followed by binding to lysosomes to form autolysosomes. The lysosomal membrane contains the lysosome-associated membrane protein (LAMP) 1 and 2. LAMP 2 is necessary for the fusion of the lysosome and an autophagic vacuole.(1) Danon disease
Danon disease is caused by mutations in the LAMP2 gene located on chromosome Xq (Nishino, 2000). It shows an XR inheritance pattern. Myopathy, hypertrophic cardiomyopathy, and mental retardation are commonly observed in male patients; whereas cardiomyopathy and associated arrhythmia (some resulting in fatal outcomes) are observed in female carriers. Cases with peripheral neuropathy have also been reported. Histopathological findings in muscle tissue are characterized by sarcoplasmic vacuoles. Dystrophin and other proteins that are usually located on the cell surface are identified on the inner surface of the vacuoles. Such vacuoles are called vacuoles with sarcolemmal- features (AVSF).
(2) X-linked myopathy with excessive autophagy (XMEA)
Myopathy with abundant autophagic vacuoles with sarcolemmal features that is transmitted as an XR trait was first reported from Finland (Kalimo, 1988). It is a slowly progressive myopathy occurring in children and adults without cardiomyopathy or mental retardation and does not affect normal life expectancy. Female carriers are asymptomatic, and it is related to mutations in the VMA21 gene located on chromosome Xq28 (Munteanu, 2008) which encodes the vacuolar membrane ATPase activity 21 protein (VMA21). VMA21 is an essential assembler chaperone that constitutes vacuolar ATPase, which acts as part of the proton pump complex of the cell. Vacuolar ATPase dysfunction affects lysosomal activity and autophagy leading to increased numbers of autophagic vacuoles. Histopathological features largely resemble those of Danon disease; however, in patients with XMEA, muscle fibers show acetylcholinesterase activity and deposition of the membrane attack complex of C5b-9 on the cell surface (Charbrol, 2001). Electron microscopy shows accumulation of debris on the cell surface between the duplicated basal lamina.
(3) Inclusion body myopathy with Paget’s disease of bone and frontotemporal dementia (IBMPFD)
IBMPFD is a hereditary multisystem proteinopathy attributable to mutations in the VCP gene that encodes the valosin-containing protein (VCP). VCP is a member of the AAA-ATPase family involved in protein degradation and cell proliferation, among other functions. IBMPFD shows an AD inheritance pattern, typically manifesting in middle age or later, as gradually progressive myopathy with proximal muscle weakness. This condition may occasionally be associated with Paget’s disease of bone, as well as dementia (Watts, 2004). Paget’s disease causes recurrent fractures with increased serum alkaline phosphatase levels. Other than IBMPFD, mutations in VCP can cause familial ALS, Charcot-Marie-Tooth disease, spastic paraplegia, and Parkinson syndrome.
Histopathological examination of muscle specimens shows moderate myopathic changes with small vacuoles including rimmed vacuoles and ragged red fibers. These myopathic changes are commonly associated with moderate neurogenic changes. Electron microscopy shows various forms of inclusions in the myonuclei and sarcoplasm (Fig. 37) (Matsubara, 2016b).
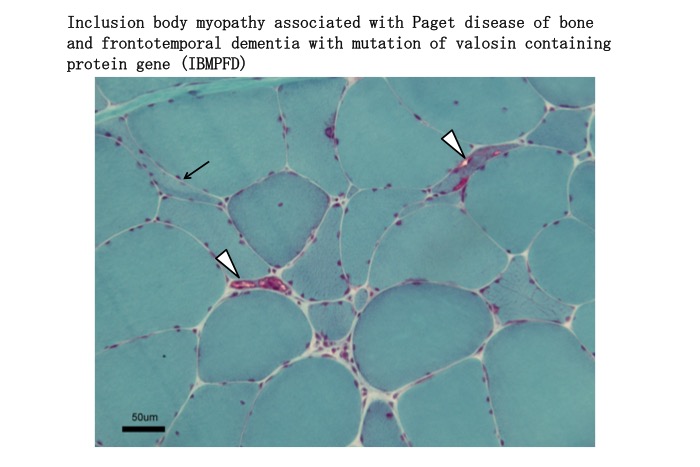
Fig.37
Muscle biopsy from a patient with IBMPFD show fibers with rimmed vacuoles (arrow heads) and scattered small angular fibers (arrow).




