Hereditary myopathies
Congenital myopathies
5. Tubular aggregate myopathy
Histopathologically, tubular aggregates appear as dark blue areas within the sarcoplasm in frozen sections stained with HE or Gomori trichrome. Immunohistochemical analysis shows high NADH-TR and low SDH activity (Fig.29).
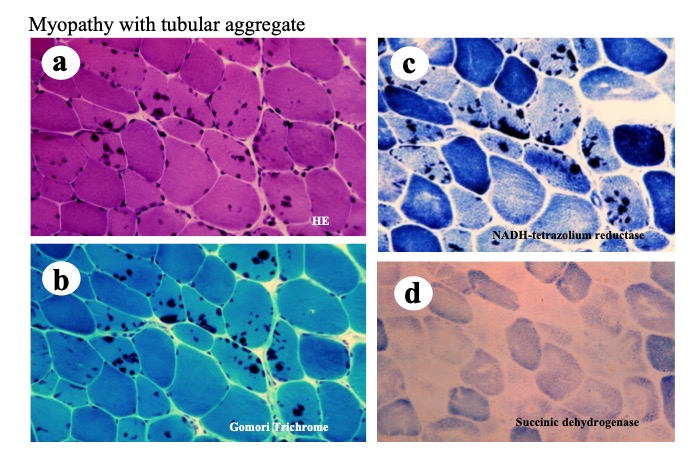
Fig.29
A muscle biopsy from a patient with familial limb girdle myasthenia (Furui, Matsubara et al. 1997). Structures stained dark with HE (a) and Gomori Trichorme (b) have strong activity of NADH-TR in the type 2 fibers (c) but no activity of succinic dehydrogenase (d), suggesting they are tubular aggregates.
Immunohistochemical studies using antibodies against the ryanodine receptor reveal high affinity of the tubular aggregates for the antibody indicating that the aggregates are derived from the sarcoplasmic reticulum (Fig.30).
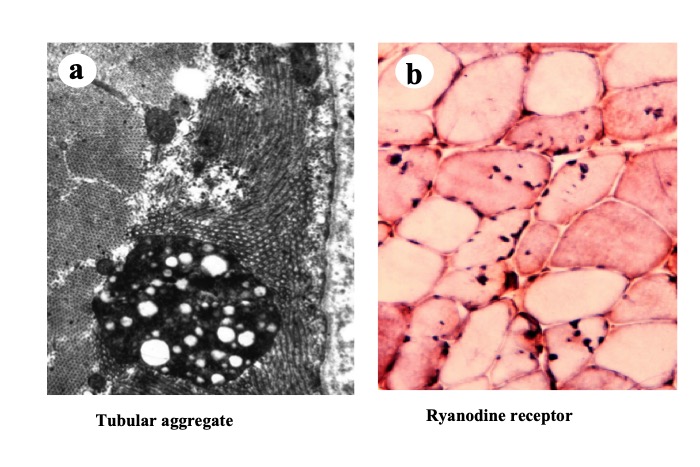
Fig.30
Electron micrograph of a tubular aggregate shows longitucinal and transvers views of tubular structure (a). Anti-ryanodine receptor antibody has affinity to the tubular aggregates (b).
Tubular aggregates are known to occur in congenital myopathies and other myopathies such as hypokalemic myopathies and hypokalemic periodic paralysis, as well as in congenital myasthenic syndrome (Furui, 1997).
Congenital myopathies with tubular aggregates are categorized into two groups as follows: (1) patients with muscle weakness that is sometimes associated with myalgia and, (2) patients with muscle weakness and myasthenia. The former group includes patients with mutations in the stromal interaction molecule 1 (STIM1) gene (Bohm, 2013) that acts as a calcium sensor on the sarcoplasmic reticulum, and mutations in the gene coding the ORAI calcium release-activated calcium modulator 1 (ORAI1), a constituent of a calcium channel that is integral to the influx and storage of extracellular calcium in response to stimulation from STIM1 (Endo, 2015). The latter group includes patients with mutations in the genes encoding enzymes involved in glycosylation of sarcoplasmic proteins. These include GFPT1 (Senderek, 2011) and DPAGT1 (Belaya, 2012).




