Hereditary myopathies
Muscular dystrophies
6. Oculopharyngeal muscular dystrophy (OPMD) and
Oculopharyngodistal myopathy (OPDM)
(1) Oculopharyngeal muscular dystrophy (OPMD)
OPMD is inherited in autosomal dominant manner. Symptoms begin in the middle age, typically with ptosis and ophthalmoplegia followed by bulbar symptoms and weakness of proximal limb muscles. Slow progress results in difficulty in walking. At early stage of disease, patients are often seen stretching their neck to look forward in order to compensate the ptosis. Hoarseness is a common initial bulbar symptom. Family pictures of patients may confirm ptosis in their family members.
An expansion of GCG repeat in the gene of polyadenylate binding protein nuclear 1(PABPN1) at 14q.11.1 was found in Canadian French and Bukhura Jewish populations (Brais, 1998, 2009). Muscle pathology shows relatively mild myopathy with occasional small angulated fibers, harboring large pale nucleus and or rimmed vacuoles. These fibers show filamentous nuclear inclusion forming network pattern. The inclusion was identified as containing PABPN1 (Tome, 1989) (Fig. 21).
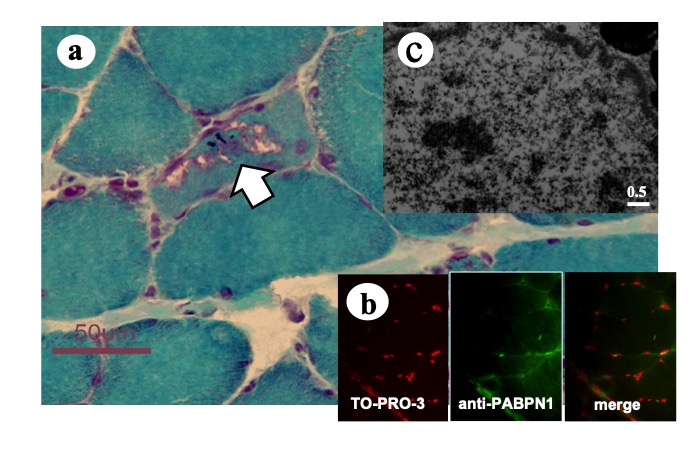
Fig.21
Oculopharyngeal muscular dystrophy (OPMD) is caused by mutation in polyA binding protein nuclear 1gene (PABPN1) in which 2 to 7 (GCG) repeats are added to the usual 6 stretch. PABPN1 shuttles between nucleus and cytoplasm. Muscle biopsy shows rimmed vacuoles (arrow) and myonuclei showing increase in PABPN1 (b). Electron microscopy demonstrates filamentous inclusions contain PABPN 1 (c).
(2) Oculopharygodistal myopathy (OPDM)
This condition is to be described here, next to OPMD, instead of in the section of congenital myopathies, because of its clinical and pathological similarity to OPMD.
Satoyoshi and Kinoshita (Satoyoshi 1977) reported from Japan an autosomal dominant hereditary myopathy with slowly progressive ptosis and extraocular palsy with weakness of the facial, masseters and bulbar muscles in association with involvement of distal limb muscles as oculopharyngodistal myopathy (OPDM). Similar condition has been reported from China, Turkey, Netherland, Italy, and the United States, but with considerable variation among them in clinical features and patterns of inheritance. Although pathological changes OPDM are like those of OPMD excepting ultrastructural features of nuclear inclusions, gene analyses showed they are different conditions. In 2019, noncoding CGG repeat expansion in LRP12 was reported (Ishiura, 2019). Later, expansion of GGC repeat was found in G1PC1 (Deng, 2020). Noncoding expansion of CGG repeat was also found in other conditions including fragile X tremor/ataxia syndrome (Hagerman, 2001), neuronal intranuclear inclusion disease (Ishiura, 2019; Sone, 2016), and coulopharyngeal myopathy with leukoencephalopathy (Ishiura, 2019).




