Hereditary myopathies
Muscular dystrophies
5. Limb-girdle muscular dystrophy
Limb-girdle muscular dystrophy (LGMD) is a diverse group of muscular disorders comprising more than 25 conditions with common symptoms and is therefore referred to as limb-girdle syndrome. The concept of this disease entity has undergone considerable revision since it was first proposed by Walton and Nattrass in 1954 (Walton, 1954) when it was characterized by the following features:
(1) onset usually late during the first decade of life or later,
(2) transmission usually as an AR but occasionally as a dominant trait,
(3) primary involvement of either the shoulder or the pelvic girdle muscles,
(4) variable rate of progression and,
(5) severe disability with inability to walk within 20 to 30 years of onset.
In 1995, the European Neuromuscular Centre workshop established the following definition of LGMD (Bushby, 1995): “Limb girdle muscular dystrophy is a genetically inherited condition that primarily affects skeletal muscle leading to progressive, predominantly proximal muscle weakness at presentation caused by a loss of muscle fibers. To be considered a form of limb girdle muscular dystrophy the condition must be described in at least two unrelated families with affected individuals achieving independent walking, must have an elevated serum creatine kinase activity, must demonstrate degenerative changes on muscle imaging over the course of the disease, and have dystrophic changes on muscle histology, ultimately leading to end-stage pathology for the most affected muscles.”
(1) Pathophysiology
The location of the related gene, the related proteins, and clinical characteristics are summarized in Table 1.
Autosomal dominant inheritance
| gene locus | mutated protein | clinical characteristics | allelic conditions and other comments | |
|---|---|---|---|---|
| LGMD1A | 5q31 | Myotilin | affects proximal muscles, nasal voice | myofibrillary myopathy |
| LGMD1B | 1q11-q21 | Lamin A/C | proximal muscles, cardiac muscles, arrhythmia | AD-EDMD, CMT2B, lipodystrophy, cardiomyopathy, progelia |
| LGMD1C | 3q25 | Caveolin 3 | proxiamal muscles, calf hypertrophy | rippling muscle disease, distal myopathy |
| LGMD1D | 7q | Desmin | upper and lower girdle muscles | |
| LGMD1E | 6q23 | DNAJB6 | proximal muscles, cardiac muscles | |
| LGMD1F | 7q32.1-q32.2 | Transportin | Spanish cases, affects proximal muscles | |
| LGMD1G | 4q21 | HNRPDL | Brazilian cases, contracture of finger joints | |
| LGMD1H | 3q25.1-p23 | unknown, located at 3p25 | Italian cases, proximal muscles, calf hypertrophy |
Autosmal recessive inheritance
| gene locus | mutated protein | clinical characteristics | allelic conditions and other comments | |
|---|---|---|---|---|
| LGMD2A | 15q15 | Calpine-3 | affects proximal muscles, winging of scapula | |
| LGMD2B | 2p13 | Dysferlin | proxiamal and distal muscles | Miyoshi myopathy, distal myopathy |
| LGMD2C | 13q12 | γ-sarcoglycan | proximal muscles, calf hypertrophy | |
| LGMD2D | 17q12-21 | α-sarcoglycan (adhalin) | proximal muscles | also called SCARMD |
| LGMD2E | 4q12 | β-sarcoglycan | proximal muscles | |
| LGMD2F | 5q33-34 | δ-sarcoglycan | proximal muscles | |
| LGMD2G | 17q11-12 | Telethonin | proximal and distal leg muscles, proximal arm muscles | |
| LGMD2H | 9q31-34 | TRIM32 | North American Hutterite cases | |
| LGMD2I | 19q13.3 | Fukutin-related protein | proximal muscles | |
| LGMD2J | 2q24.3 | Titin | Finnishi cases, anterior tibial muscle | myofibrillar myopathy |
| LGMD2K | 9q34.1 | POMT | Turkish cases, early onset, mental impairment | |
| LGMD2L | 11p13-p12 | Anoctamin 5 | French canadian cases, proximal muscles | |
| LGMD2M | 9q31 | Fukutin | Japanese cases, early onset | Fukuyama congenital muscular dystrophy |
| LGMD2N | 14q24 | POMT2 | hypotonia, delayed motor development, winging scapula | α-dystroglyconopathywith mental retardation (CMDB2) |
| LGMD2O | 1p32 | POMGnT1 | early onset, proximal muscles, winging scapula | |
| LGMD2P | 3p21 | α-dystrooglycan (DAG1) | early onset, proximal muscles, mental impairment | |
| LGMD2Q | 8q24 | Plectin | delayed motor development | CMD with familial junctional epidermolysisbullosa |
| LGMD2R | 2q35 | Desmin | facial, proximal limb, respiratory and cardiac muscles | |
| LGMD2S | 4q35.1 | TRAPPC 11 | facial and proximal muscles, mentaly impaired |
Tab.1
Limb-girdle muscular dystrophy
(2) Common histopathological changes
Histopathological changes differ considerably depending on the types, although most types share the following features:
(a) They all show changes indicating chronic myopathy (Fig. 18).
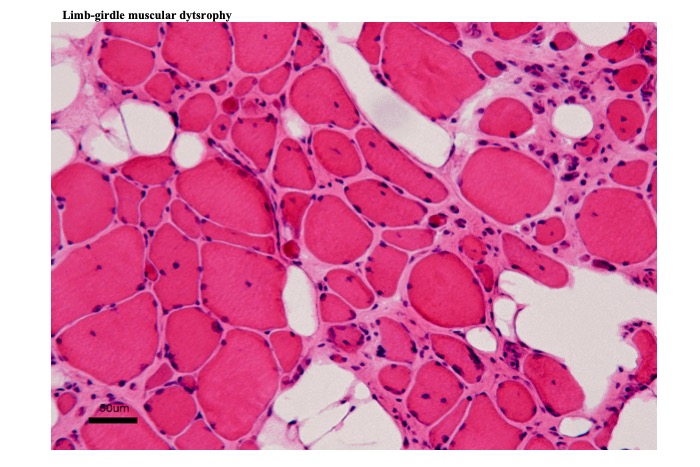
Fig.18
Muscle biopsy shows changes of chronic myopathy. Increased variations in muscle fiber diameter and frequency of internal nuclei. Contour of fibers are round. Interstitial tissue is extended with fibrosis. Empty vacuoles in the interstitial tissue represent deposition of fatty tissue.
(b) Increased variations in fiber diameter secondary to combinations of atrophic and hypertrophic fibers.
(c) The hypertrophic fibers often show fiber splitting.
(d) Internal nuclei are very commonly observed, and split fibers often show nuclei at one end of the rift.
(e) Various degrees of fibrosis and fat deposition occur in the interstitial tissue.
(3) Clinical symptoms and histopathological findings in each type of LGMD
i) LGMD1A
Myotilin (MYOT) or myofibrillar titin-like immunoglobulin domain protein (TTID) is a structural protein present in the Z line that ensures sarcomere integrity by maintaining strong linkage between actin filaments and other proteins that constitute the Z line. Mutations in the TTID (or MYOT) gene cause LGMD1A and myofibrillar myopathy (Salmikangas, 1999; Selcen, 2004). LGMD1A manifests after the age of 40 and causes distal or proximal muscle weakness or muscle pain with exercise.
Weakness gradually worsens and occasionally results in respiratory failure. The vocal cords and pharyngeal muscles can be affected in a few patients with distal muscle weakness (Salmikangas, 1999). Cardiac muscles and peripheral nerves can also be affected (Olive, 2005).
Histopathological changes in LGMD1A include features of myofibrillar myopathy accompanied by vacuole formation (rimmed vacuoles in a few cases) (Carisson, 2007). Although an increase in the quantity of MYOT is reported, this finding may also be observed in patients with nemaline myopathy and central core disease (Schroder et al., 2003).
ii) LGMD1B
Lamin A/C is an important structural protein component of the nuclear envelope. This nucleoskeletal protein stabilizes the shape of the nucleus and controls protein synthesis. Mutations in the LMNA gene on chromosome 1q11-23 cause a wide variety of disorders including LGMD1B (Kitaguchi, 2001), ADEDMD (Bonne, 1999), dilated cardiomyopathy, cardiac conduction block (Sinagra, 2001), Dunnigan type familial partial lipodystrophy (Cao, 2000), mandibuloacral dysplasia (Novelli, 2002), Hutchinson-Gilford progeria syndrome (Kirschner, 2005), and AD axonal Charcot-Marie-Tooth disease type 2C (Goizet, 1967) among others.
Myopathological changes in LGMD1B correspond to those noted in cases of relatively moderate myopathy. Ultrastructural studies reveal changes in the nuclear envelope (Matsubara, 2004). Notably, routine immunohistochemical methods might not reveal any abnormality.
iii) LGMD1C
Caveolin 3 is a protein that is expressed exclusively in muscle cells. It forms the caveolae-sarcolemmal invaginations measuring 50–100 nm in diameter, in the plasma membrane of the muscle fiber. It plays an important role in transportation of materials through the plasma membrane and in signal transduction. It is closely related to dysferlin and other membrane proteins, which collectively play a role in the maintenance of the plasma membrane and the T system. Mutations in the CAV3 gene located on chromosome 3p25 cause LGMD1C (Minetti, 1998), as well as idiopathic hyperCKemia and rippling muscle disease (Betz, 2001).
Clinically, LGMD1C usually manifests in childhood and causes weakness and spasm of proximal or distal muscles. Rippling muscle disease is associated with increased excitability of muscle resulting in ripple-like (wave-like) self-propagating muscle contraction in response to light percussion on the muscle surface with peripheral spread of the ripple. Cardiomyopathy is another feature associated with CAV3 gene mutations (Gazzerro et al., 2010).
In addition to moderate myopathic changes, immunohistochemical studies reveal absence or reduction in the quantity of caveolin 3 on the surface of the muscle fibers. Moreover, secondary reduction of caveolin 3 expression has been reported in patients with LGMD2B.
Lipodystrophy along with clinical features of LGMD1C has been reported in cases showing gene mutations in the caveolin-associated protein polymerase I and transcript release factor (PTRF, also known as Cavin) (Hayashi, 2009).
iv) LGMD1D, E, F, G, H
Mutations of the gene DNAJB6 at 7q36 which encode heat shock protein (HSP) 40 was identified in the patients with pathology of muscular dystrophy and myofibrillar myopathy, showing rimmed vacuoles and deposition of TDP43 (Sarparanta, 2012). This rare condition was initially reported as LGMD1D . International Workshop of European Neuromuscular Conference (ENMC) proposed a new name for this condition, LGMD D1 DNAJB6-related myofibrillar myopathy (Straub, 2018). Myopathologically it often shows sarcoplasmic masses and ring fibers as well as lobulated fibers as demonstrated in the virtual slide. New classification of LGMD will be adopted in this page when it is commonly used.
LGMD 1E were originally reported as a dominantly inherited myopathy showing changes compatible with myofibrillar myopathy. Clinically distal weakness and cardiomyopathy was described.
A frame-shift mutation in the gene of transportin 3 (TNPO3) at 7q32 was identified in an Italo-Spanish family with VLGMD1F (Torella 2013; Melia, 2013).
LGMD1G causes slowly progressive weakness of late onset (Sterling, 2004). Mutation in the gene of heterogeneous nuclear ribonucleoprotein D-like protein (HNRPDL) at 4q21 was reported (Vieira, 2014). The protein plays a role in RNA production.
Slowly progressive proximal weakness is the main symptom of LGMD1H. Related gene was located at 3p23-p25.1 (Bisceglia, 2010).
v) LGMD2A
Diseases classified as LGMD2 show an AR inheritance pattern. LGMD2A occurs following mutations in the CAPN3 gene located on chromosome 15p15.1, which encodes calpain-3 (CAPN3). CAPN3 is a calcium-dependent protease that is categorized as a sarcoplasmic protein but may be localized in the nucleus. The exact pathomechanism by which lack of CAPN3 causes muscle fiber degeneration is unclear; however, it is hypothesized that CAPN3 activates a substrate that is stored in an inactive state when this substance is needed for maintenance of the muscle fiber.
Disease onset typically occurs between ages 10 and 15, although considerable variations are observed. It manifests with weakness of the hip girdle muscles, particularly the hip adductors and the gluteus maximus. The abductors (gluteus minimus and medius and the piriformis) are less affected. It causes a waddling gait and difficulty in running and climbing stairs. Winging of the scapula (scapula alata) may also occur. Leg weakness progresses gradually causing shortening of the Achilles tendon and although obvious changes are infrequent, a moderate degree of pseudohypertrophy of the calves may occur. Serum CK levels are elevated. The severity of the disease varies; however, many patients are wheelchair-bound by approximately 30 years of age. Cardiomyopathy is rare, and mental function remains intact (Fardeau, 1996).
Histopathological examination of muscle specimens shows myopathy with type 1 fiber predominance and frequent lobulated fibers. Immunohistochemical studies reveal normal appearance of dystrophin and sarcoglycans. Although CAPN3 expression can be identified immunohistochemically, owing to the unstable nature of the protein, it is difficult to use this as a reliable marker and it is therefore not considered in the routine examination. Immunoblotting is preferred but is susceptible to secondary reduction in other myopathies.
vi) LGMD2B
Dysferlinopathy is caused by mutations in the DYSF gene, which codes dysferlin and shows diverse clinical features including those of LGMD and Miyoshi myopathy with distal muscle weakness and muscle atrophy. They may not represent different disease entities but are components of a wide spectrum of symptoms of the same disease. Patients show significantly high serum CK levels in their teens with mild evidence of muscle weakness, and the condition may often present as idiopathic hyper-CKemia. However, later in their 20s, leg weakness becomes evident. Distal muscles tend to be thin, and calf hypertrophy is not observed even in patients with predominantly proximal weakness. Progression of weakness varies greatly, and cases of late-onset disease have been reported (Kling, 2008).
Histopathological findings correspond to those identified in cases of myopathy, and only mild changes are obvious in the early stages. Inflammatory changes must be distinguished from inflammatory myopathies (Fig. 19).
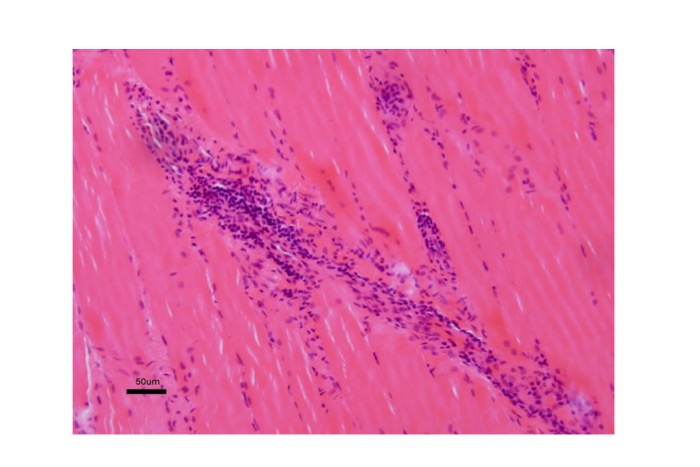
Fig.19
LGMD2B : Inflammatory change can be seen in dysferlinopathy mimicking myositis.
No differences are observed between LGMD and Miyoshi myopathy. Immunohistochemical studies show absence of dysferlin (Fig. 20).
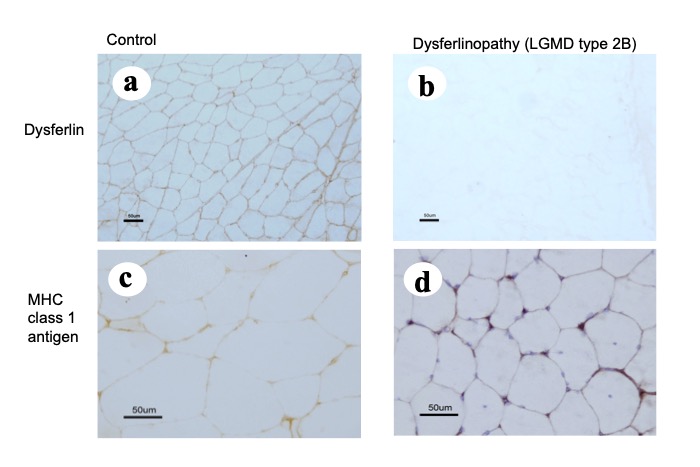
Fig.20
In LGMD2B (dysferlinopathy), expression of dysferlin is not seen (b), and a moderate degree of aberrant expression of MCH class I antigen can be seen (d).
However, a low affinity for the antidysferlin antibody is commonly observed in other muscle diseases including muscular dystrophies. Genetic analysis is recommended for diagnostic confirmation.
Identical mutations are observed in cases of LGMG2B and Miyoshi myopathy. Both types of distribution of weakness (proximal and distal) can occur in patients with the same mutation (Illarioshkin, 2000). Genetic analyses reported by previous studies showed compound heterozygotes in 66% and one pathological allele in 22% of cases (Krahn, 2009). A decrease in dysferlin in carriers has also been reported (Famin, 2006).
vii) LGMD2C, D, E, F
Sarcoglycans form a sarcolemmal dystrophin-associated glycoprotein complex, which binds the subsarcolemmal cytoskeletal proteins and sarcolemma. The four main types of sarcoglycans include α, β, γ, and δ. Sarcoglycanopathies refer to a group of muscle-wasting disorders caused by genetic defects in one of the four sarcoglycans and often present with pseudohypertrophy of the calves (Eymard, 1977) and macroglossia, similar to features observed in dystrophinopathies.
LGMD2C or γ-sarcoglycanopathy, also called adhalinopathy is observed in approximately 2% of patients with muscular dystrophies in Japan (Hayashi, 1955). It was originally designated as severe childhood AR muscular dystrophy (SCARMD) based on the relatively severe clinical symptoms, which resemble those associated with dystrophinopathies. Histopathological changes in muscle specimens are more severe than those observed in cases of LGMD2A and B. Necrosis and regeneration of muscle fibers are common, whereas lobulated fibers are relatively uncommon, and only few type 1 fibers are identified. Immunohistochemical studies show lack of affinity for the anti-adhalin antibody in 53% of cases and a reduced affinity in 47% (Eymard, 1955).
Clinical manifestations of LGMD2D (α-sarcoglycanopathy), LGMD2E (β-sarcoglyconopathy), and LGMD2F (δ-sarcoglyconopathy) are similar. All categories usually show disease onset in childhood weakness of hip girdle muscles leading to a waddling gait, a positive Gowers’ sign, and difficulty in running. Some children may show exercise intolerance or muscle cramps. These symptoms are invariably progressive, and patients may be unable to walk in adulthood. Histochemical examination and immunoblotting using a panel of anti-sarcoglycan antibodies are used for diagnosis. However, sarcoglycans act collectively as a complex; therefore, mutations in one category of sarcoglycan cause a reduction in other groups. Although this cannot be conclusively established, it is usually accepted that the mutated variety shows the most significant reduction.
viii) LGMD2 G, H, I, J, K, L, M, O
LGMD2G constitutes approximately 2.7% of AR LGMD. It is caused by mutations in the teletonin (TCAP) gene (Vanizof, 2002). Teletonin is a 19-KD protein present in the Z line and anchors titin to the Z line. Clinically, LGMD2G causes proximal or distal muscle atrophy with winging of the scapula and calf hypertrophy. It may cause cardiomyopathy (Hayashi, 2004) and foot drop. Immunohistochemical examination reveals absent or reduced affinity for the anti-teletonin antibody (Francis, 2014)
LGMD2H is a rare condition first reported in 1976 in the Canadian Hutterite population. The causative genetic mutation was identified on chromosome 9q31-33 in 1998. It was identified as a point mutation within the NHL domain of tripartite motif-containing 32 protein (TRIM32) gene (Frosk, 2002). TRIM32 is an E3-ubiquitin ligase, and the NHL domain is considered the locus where TRIM32 binds to the target protein for ubiquitination. Mutations in TRIM32 leading to sarcotubular myopathy have been reported in the American Hutterite population (Jerusalem, 1973) and in south Germany. LGMD2H causes gradually progressive proximal muscle weakness and muscle atrophy and is often associated with facial muscle atrophy, scapular winging, calf hypertrophy, and ankle contractures (Shieh, 2011) and may occasionally show facioscapulohumeral distribution. Serum CK levels are moderately elevated and nEMG reveals a mixed pattern of myopathic and neurogenic changes. Myopathologically, myopathic changes may be associated with features of vacuolar myopathy and fiber-type grouping. Some carriers show vacuolar myopathy.
LDMD2J is a rare condition secondary to mutations in the TTN gene, which encodes titin in the Z-line and results in myofibrillar myopathy.
LDMD2I, K and M are caused by mutations in fukutin-related protein (FKRP), protein-O-mannosyl transferase 1 (POMT1), and the fukutin (FKTN) genes, respectively. These proteins play an important role in glycosylation of α-dystroglycan. Dysfunction of these proteins results in dysplasia of the basal lamina of the muscle fibers causing muscle fragility. This group of conditions is referred to as α-dystroglycanopathy (Muntoni, 2011). LGMD2I is uncommon in Asians but perhaps constitutes the most common type of LGMD in Europe. It manifests in childhood at approximately a mean age of 9 years and presents with easy fatigability or myalgia after exercise. Patients develop progressive weakness of the proximal muscles of the lower limbs, followed by symptoms in the upper limbs. Winging of the scapula is occasionally observed. Patients may be wheelchair-bound by 20 years of age. Respiratory failure may occur at a mean age of 16 years after disease onset (Bourteel, 2009). Histopathological examination of muscle tissue reveals findings corresponding to myopathic changes. Immunohistochemical examination reveals reduced α-dystroglycan expression.
POMT1 gene mutations have been identified as a cause of Walker-Warburg syndrome (WWS), a severe form of congenital muscular dystrophy. LGMD2K is reportedly a milder form of WWS (Dincer, 2003; Balci, 2005).
Mutations of the FKTN gene were originally identified in the Fukuyama type of congenital muscular dystrophy (FCMD). LGMD2M is characterized by lack of mental retardation and dysplasia of the cerebral cortex (which are common in FCMD) (Murakami, 2006; Godfrey, 2006).
LDMD2K and LGMD2M will be discussed in detail in the section describing congenital muscular dystrophy.
A few papers have described LGMD2P and LGMD2Q.




