Hereditary myopathies
Congenital myopathies
Congenital myopathy was originally defined as a group of hereditary myopathies with relatively slow progression and benign nature compared to muscular dystrophies. However, as an increasing number of cases were reported for each subgroup of congenital myopathy, particularly after technical advances that enabled genetic analyses for diagnostic purposes, it was discovered that patients could show various rates of progression and severity. Therefore, the boundary between congenital myopathy and muscular dystrophy has become blurred.
1. Nemaline myopathy
Nemaline bodies are small spindle-shaped rods or short thread-like structures primarily distributed in the sarcoplasm. They stain dark purple with Gomori-trichrome (Fig. 25).
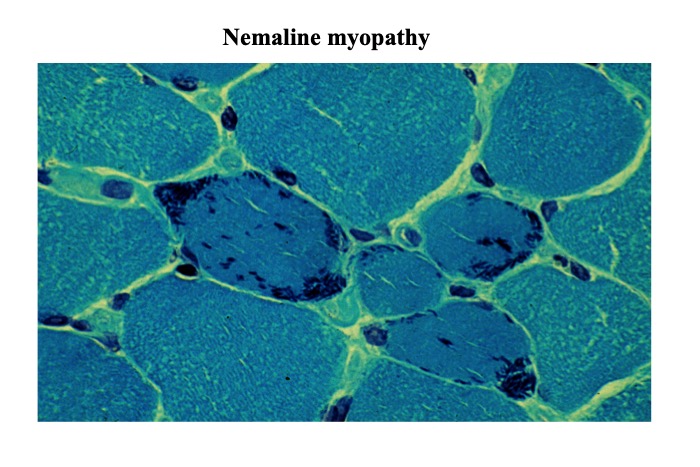
Fig.25
Using electron microscopy, they appear as spindle-shaped electron-dense structures with electron density similar to that of the Zdisks. In fact, they are often observed in continuation with the Z-disk (Fig. 26).
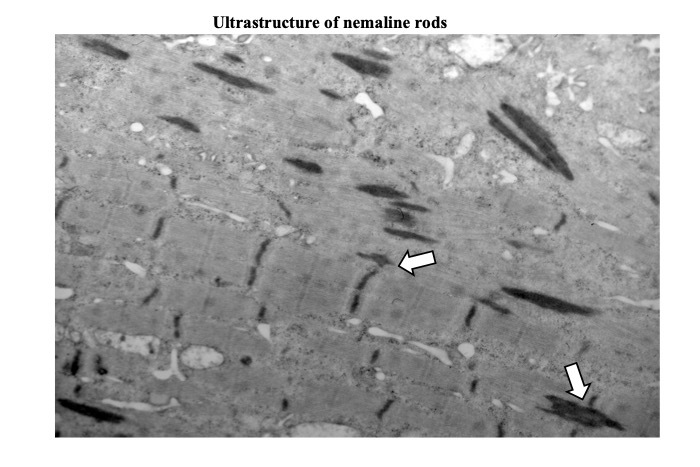
Fig.26
Nemaline rods are often seen in close proximity to the Z disks (arrows).
Nemaline myopathy refers to congenital myopathy characterized by the presence of nemaline rods. However, careful evaluation is necessary to diagnose nemaline myopathy, because the detection of nemaline rods is a nonspecific change as they can appear in many other conditions including muscle dystrophies and inflammatory myopathies. Additionally, nemaline myopathy is not a single disease, and at least seven different conditions have been identified as subgroups. Therefore, “nemaline myopathies” is the appropriate nomenclature to describe these conditions.
Approximately 50% of the patients with nemaline myopathy show mutations in the neblin (NEB) gene. Neblin is an important constituent of the actin filament. The condition shows an AR inheritance pattern, and the clinical presentation ranges between benign and severe forms. The typical congenital form is classified under the latter variety.
Patients with mutations in skeletal α-actin 1 gene (ACTA1) constitute approximately 20% of all patients with nemaline myopathies. Although severity may vary, patients commonly present with a severe presentation. Symptom usually manifest in childhood. The condition shows an AD inheritance pattern.
The Kelch-like family member (KLHL) 40 gene mutation occurs in approximately 5% of cases. Mutations in KLHL 40 and 41 genes usually manifest at birth. The condition shows an AR inheritance pattern.
Patients with mutations in tropomyosin 3 and 4 (TPM) genes account for approximately 5% each of all cases. The presentation is relatively mild in severity, and the condition shows an AR inheritance pattern.
Patients with mutations in the RYR1 gene show an AR inheritance pattern.
A small number of cases with mutations in the leimodin (LMOD) 3 gene have been reported. This condition shows an AR inheritance pattern.
Nemaline and congenital myopathies typically present with weakness and atrophy of the muscles of the face and neck and the proximal muscles of the limbs. However, distal muscles may be involved first in a small number of cases. In addition to the facial appearance that characterizes myopathic conditions, patients show a tent-shaped upper lip, high-arched palate, and a retracted jaw. In benign cases, patients may be able to walk in childhood, although respiratory failure develops in later life. Serum CK levels remain normal.
Among adults with nemaline myopathy, those who present with characteristic facial features are classified as patients with congenital nemaline myopathy of late onset. However, a few patients lack features that characterize the congenital form. Among this latter group, patients with myopathy of acute onset and progression are often known to show M-protein in the serum (Engel, 1966b). Some patients respond to immunotherapy including corticosteroids, immunosuppressants, and autologous peripheral blood stem-cell transplant (Doppler, 2013). These patients include those with neurogenic changes or patients with human immunodeficiency virus (HIV) infection.




