Hereditary myopathies
Muscular dystrophies
8. Congenital Muscular Dystrophy
Congenital muscular dystrophy (CMD) was originally understood as muscular dystrophy that manifests at birth. However, with advances in molecular biology, the distinction between congenital myopathies and CMD has become blurred. Moreover, many types of LGMD show early onset similar to that observed in CMD. Currently, only a few conditions are classified as CMD and include cases with severe muscular symptoms associated with a variety of ocular, neurological, and other complications. In this section, we discuss four diseases based on the American Academy of Neurology classification.
(1) Collagen type VI myopathies
Collagen constitutes the major structural component of the interstitium in muscle fibers. Ullrich CMD and Bethlem myopathy are caused by mutations of genes encoding the collagen type VI proteins (COL6A1, COL6A2, COL6A3). Although they were originally described as two different conditions, a greater accumulation of cases has led to the understanding that they are members of a group of allelic diseases caused by mutations of the COL6A genes. Moreover, conditions with a clinical phenotype of LGMD and AR myosclerosis have also been reported.
Ullrich CMD is a severe condition with contractures of joints of the lower limbs and is often associated with hyperlaxity of the finger joints. Muscle weakness and scoliosis appear later during the disease course with concomitant hyperkeratosis and keloid formation. Many patients cannot walk and later develop respiratory insufficiency. Both AD and AR inheritance patterns are known. Serum CK levels remain normal or are minimally increased.
Compared to Ullrich CMD, Bethlem myopathy is a less severe condition and usually shows an AD inheritance pattern, although a small number of families have shown recessive inheritance. Contracture of the long finger flexors is noted. Children develop contractures of the ankles concomitant with weakness of proximal muscles and contracture of the elbows at approximately 10 years of age. Muscle weakness is often progressive, and patients may experience difficulty walking in their 60s. Some patients may develop respiratory failure. Serum CK levels remain normal or are slightly increased.
Collagen VI is essential for the development and maintenance of muscle fibers, forming microfibrillar network with the basal lamina of the muscle cells. Mutations of the COL6A gene cause various abnormalities in muscles, as well as in tendons, cartilage, skin, and vertebral disks.
Myopathological features of collagen type VI myopathies resemble those of non-specific myopathy. Ullrich CMD presents with a disproportionate distribution of the fiber types with predominance and atrophy of type 1 fibers (Schessl, 2008). AR cases of Ullrich CMD with negative staining for collagen VI have been reported (Higuchi, 2001). In contrast, AD cases show immunopositivity for collagen VI, although double staining with basal lamina protein shows abnormal distribution of collagen VI (Pan, 2003). This finding is attributable to differences in form and location of mutations in the COL6A gene.
(2) Merosin (lamininα2) and integrin α7 deficiency
Laminins are proteins that form the basal lamina. Merosin (laminin α2) is the most abundant of the 12 varieties of laminins occurring in muscle tissue. It is a heterotrimer of α2-β1-γ1 chains. Merosindeficient CMD (MDC1A) is common in Europe. Patients show hypotonia at birth with muscle weakness, multiple contractures of the joints, and respiratory failure. However, no cardiomyopathy and mental retardation are identified. Histopathological examination of muscle tissue reveals dystrophic changes.
Type 1 fiber atrophy is common, and lamininα2 is undetectable on the surface of muscle fibers (Tome, 1994). However, laminin α2 can be difficult to detect even in cases of FCMD, which will be described in the next section.
Integrinα7 is a ligand of merosin occurring on the surface of the muscle fiber along withα dystroglycan. This protein also supports the basal lamina. Patients show hypotonia at birth and delayed developmental motor milestones in addition to mental retardation. Serum CK levels are minimally elevated (Hayashi, 1998). Mutations in the ITGA7 gene and reduction of labeling by antibody have been reported (Pegoraro, 2002).
(3) α-dystroglycanopathies
The extracellular peripheral glycoprotein α-dystroglycan plays a pivotal role in the formation and maintenance of the surface structure of the muscle fiber. This protein undergoes post-translational modification including glycosylation and mannosylation. Mutations in the genes encoding enzymes involved in the post-translational modification cause several CMDs.
i) Fukuyama congenital muscular dystrophy
Most cases of CMD observed in Japan are categorized as Fukuyama congenital muscular dystrophy (FCMD) and most patients are Japanese. Muscle atrophy and contracture of multiple joints are noted at birth along with mental retardation. Impairment of eye movements, cortical blindness and other ophthalmological symptoms occur in approximately 50% of patients. Following the accumulation of a greater number of cases in the literature since the original genetic diagnosis, a greater number of patients with relatively mild symptoms and slow progression are now recognized as showing this condition.
FCMD is related to abnormalities of the fukutin protein, which is encoded by the FKTN gene located on chromosome 9q31. Many patients show insertion of a 3 kb retrotransposon in a non-coding region of the FKTN gene. It is concluded that this mutation is inherited from a founder in ancient Japan (founder effect). Most patients are homozygotes for retrotransposon insertion. However, other mutations including deletion and nonsense mutations are also known to occur. A small number of compound heterozygotes for these mutations have been observed (Toda, 1999). Presumably, fukutin is involved in the glycosylation of α-dystroglycan (Kobayashi, 2001; Yamamoto, 2010).
Histopathological examination of muscle tissue in cases of FCMD shows findings similar to those observed in cases of muscular dystrophy. Immunohistochemical studies show decreased quantities of lamininα2, which is an important constituent of the basal lamina, with irregularity and disruption of the protein, as well as the basal lamina (Fig. 24).
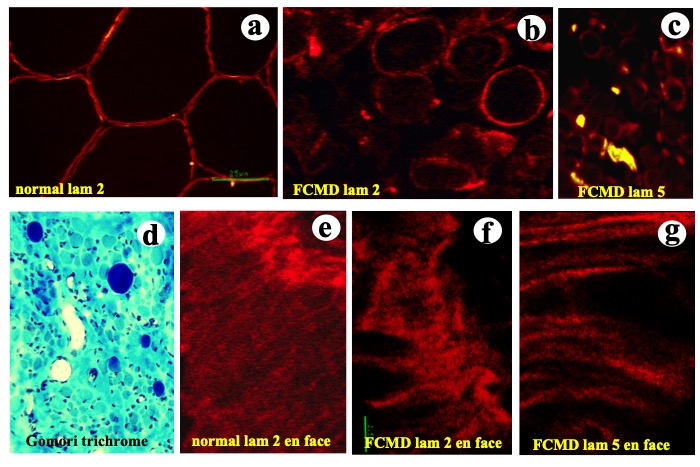
Fig.24
Laminins are major components of the basal lamina. Laminin 2 stains all around the muscle fiber in normal muscle (a). Fukuyama congenital muscular dystrophy (FCMD) shows myopathic changes (d), while lamlaminins 2 and 5 show disruptions (b,c). En face views of the cell surface observed with confocal laser microscope show intact laminin 2 in normal muscle (e) whereas FCMD muscle shows disruption of laminins 2 and 5 (f.g.).
Electron microscopy confirms the diagnosis. The plasma membrane of the muscle cell shows degeneration at places corresponding to loss of the basal lamina (Matsubara, 1999).
ii) Muscle eye brain disease
Muscle eye brain disease (MEB) was first reported from Finland and was attributed to mutations in the POMGnT1 gene, which encodes a protein named glycosyltransferase, protein O-mannose β-1,2-Nacetylglucoseaminyltransferase 1(POMTGnT1). MEB shows a wide variety of clinical manifestations and a global presence (Taniguchi, 2003). Most patients present as “floppy neonates” and are unable to walk later. Visual disturbances occur secondary to severe myopia, optic nerve hypoplasia, retinal detachment, or retinitis pigmentosa in addition to mental retardation and epilepsy. Cerebral and cerebellar dysplasia are also noted. Reduced activity of the POMTGnT1 enzyme is reported. Histopathological findings in patients with MBE are similar to those in patients with α-dystroglycanopathies. Genetic studies are needed for diagnostic confirmation.
iii) Walker-Warburg syndrome
Walker-Warburg syndrome (WWS) is a severe form of CMD with a variety of central nervous system malformations, such as cortical dysplasia, lissencephaly, absence of the corpus callosum, severe hydrocephalus, and arachnoid cysts. Patients usually live only for a few years. Previous studies have confirmed that WWS differs from other α-dystroglycanopathies and is attributable to mutations in the protein O-mannosyltransferase 1 (POMT1) gene involved in mannosylation of α-dystroglycan. Histopathological findings resemble those observed in α-dystroglycanopathies. Hydrocephalus and arachnoid cysts are common in cases of WWS. Genetic analysis is needed for diagnostic confirmation.
iv) Otherα-dystroglycanopathies
Following are the other α-dystroglycanopathies reported in the literature:
Mutations in the fukutin-related protein (FKPP) (FKPP) gene cause congenital muscular dystrophy 1C (MDC1C) and LGMD 2I (LGMD2I). FKPP occurs in the Golgi complex and is involved in glycosylation of α-dystroglycan. Patients with MDC1C show a variety of symptoms. It can show the clinical presentation of MEB or WWS. Central nervous system and cardiac muscle involvement is known to occur.
The Large gene located on chromosome 22q12 is the 5th largest gene in the human genome. The Large protein is also considered a α-glycosyltransferase. It was originally discovered in a dystrophic mouse and later in humans with MDC1D (Longman, 2003).
Histopathological findings in muscle specimens obtained from patients with MDC1C and MDC1D include dystrophic changes. Using antibodies against the glycosylation-epitope of the α-dystroglycan, Jimenez-Mallebrera (2009) demonstrated reduced staining of α-dystroglycan in patients with mutations in the POMT1, POMT2 and POMTGnT1 genes; however, these findings were not observed in patients with mutations in the FKPP and Large genes.
(4) Rigid spine muscular dystrophy and multi-minicore myopathies
Spinal rigidity is not a specific symptom but can occur in several conditions including AD-EDMD secondary to mutations in the LMNA gene. However, CMD associated with spinal rigidity was first described as an independent entity by Dubowitz et al. in 1973. Whether it was “a syndrome in search of a name” was clarified following the discovery of mutations in the selenoprotein N1 (SEPN1) gene located on chromosome 1q36 (Moghadaszadeh, 1998, 1999) in this condition, which was later named rigid spine muscular dystrophy (RSMD1). Selenoprotein is a glycoprotein localized to the endoplasmic reticulum and is involved in the oxidative stress response and calcium homeostasis. It is significantly expressed in fetal tissues and is considered to play a role in muscle development. Mutations in SEPN1 lead to myopathy with the presence of Mallory bodies. The term “SEPN-related myopathy” was proposed by Ferreiro in 2004.
The clinical presentation includes poorly developed musculature, kyphoscoliosis, and respiratory failure in severe cases. Histopathological findings show a mild degree of myopathic change that is occasionally associated with uneven distribution of oxidative enzymes, resulting in minicore formation.
Multicore disease was first described by A. G. Engle et al. (1971) as a benign non-progressive myopathy in children. Histopathologically, it is characterized by multiple small cores devoid of mitochondria distributed in the sarcoplasm. Subsequently, other studies documented multiminicore myopathy as a concomitant feature of several conditions that occur in association with mutations in the SEPN1, RYR1, and myosin heavy chain 7 (MYH7) genes (Cullup, 2012) among others. Therefore, it is appropriate to refer to these disorders as multiminicore myopathies.




