Hereditary myopathies
Metabolic myopathies
1. Abnormal glycogen metabolism (Glycogen storage diseases)
Abnormalities of the enzymes involved in glycolysis interfere with adequate energy supply to cells. Blockage of normal metabolic pathways leads to the accumulation of several products that cause cellular injury. Among the several glycogen storage diseases reported, followings cause clinically manifest myopathy.
(1) Pompe disease (Glycogen storage disease type II)
The lysosomal enzyme acidαglucosidase (GAA or acid maltase) catabolizes glycogen to glucose. Pompe disease is caused by a deficiency of GAA consequent to mutations of the GAA gene. It shows an AR inheritance pattern and presents as infantile, juvenile, or adult forms. Patients with the infantile form are born as floppy infants with respiratory and heart failure secondary to cardiomyopathy. It is a classical form with early mortality, usually within the first year of life, in untreated cases. Symptoms of myopathy predominate in the juvenile and adult forms. Respiratory failure may be prominent, although heart failure is relatively uncommon. Hyper-CKemia is usually present, and this condition is often misdiagnosed as LGMD. Awareness regarding this condition has increased in recent years following the development of effective enzyme replacement therapy.
Histopathological examination of muscle tissue shows myopathic changes with vacuoles. Using PAS reaction, several fibers show intense staining of vacuoles, as well as the sarcoplasm. However, many vacuoles appear empty as their contents are lost during tissue preparation. This particularly occurs with paraffin-embedded sections. Histochemical examination reveals increased activity of acid phosphatase in the sarcoplasm. Plastic (EPON)-embedded sections tend to preserve vacuolar contents. Electron microscopy reveals accumulation of glycogen granules both in the sarcoplasm and in the membranebound vacuoles (Fig. 35).
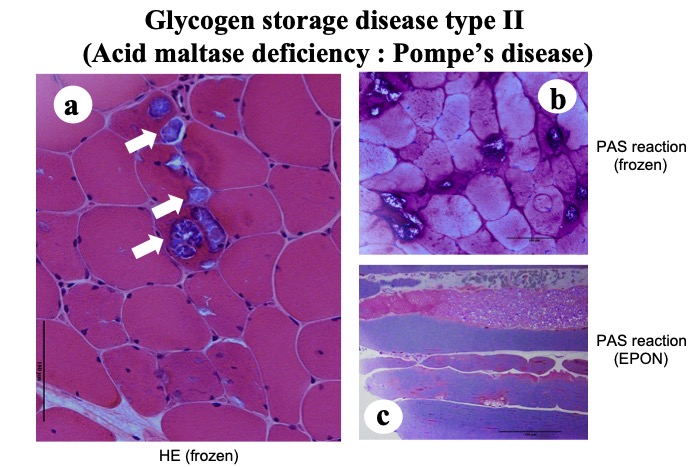
Fig.35
A case of Pompe disease of adult form. HE shows fibers with vacuoles containing granular material (a: arrows). Both frozen and EPON sections (b,c) stained with PAS reaction show positive material both in the vacuoles and in the sarcoplasm.
Rarer forms of glycogen storage disease with myopathic changes include the debrancher enzyme deficiency (glycogen storage disease type III) and the branching enzyme deficiency (glycogen storage disease type IV), and both conditions present with hepatomegaly.
(2) McArdle disease (Glycogen storage disease type V)
McArdle disease is caused by absence of myophosphorylase, which is involved in the degradation of glycogen to glucose 1 phosphate (G1P) for optimal control of glycolysis. It occurs secondary to mutations in the PYGM gene and shows an AR inheritance pattern. Clinically, it presents with easy fatigability, exercise-induced muscle pain, muscle cramps, and transient weakness, all of which are usually ameliorated by brief rest. The “second wind” phenomenon refers to amelioration of these symptoms despite continuation of exercise or after a brief period of rest, and this phenomenon may provide a clue to the diagnosis. Muscle weakness is not obvious at rest. A history of episodic dark reddish colored urine suggesting hemoglobinuria may be positive. Myopathologically, abnormal accumulation of glycogen is observed in the muscle cells; however, minimal changes indicative of muscle fiber destruction are noted. Absence of phosphorylase activity on muscle biopsy confirms the diagnosis.
(3) Tarui disease (Glycogen storage disease type VII)
During glycolysis, phosphofructokinase (PFK) catalyzes the ATP-dependent phosphorylation to convert fructose-6-phosphate to fructose-1,6-diphosphate. Among the three isozymes of PFK, the muscle-type isoenzyme is present in muscles and erythrocytes. This enzyme is absent in patients with Tarui disease who present with symptoms resembling those of McArdle disease, except that patients with Tarui disease develop hemolytic anemia and hyperuricemia, and the “second wind” phenomenon is rare. Fixed muscle weakness is known to occur in some cases (Malfatti, 2012). Histopathological findings in muscle tissue include moderate myopathy with accumulation of glycogen within vacuoles. Polyglucosan bodies are identified in a few cases (Malfatti, 2012). Histochemical analysis confirms the absence of PFK.




