Neurogenic muscular atrophy
Hereditary muscular atrophy
1. Spinal muscular atrophy
Spinal muscular atrophy (SMA) is a disorder showing an autosomal recessive (AR) inheritance pattern. Based on its severity, it is classified into four clinical subtypes. Type I SMA or Werdnig-Hoffmann disease is the most severe form in which symptom onset occurs in utero or during early life, and patients cannot sit up unaided. Patients with type II or intermediate SMA can sit up unaided but can never ambulate. Type III SMA or Kugelberg-Welander syndrome manifests later in life, and patients can ambulate. Main symptoms include proximal muscle weakness, often associated with fasciculations and tremor. Type IV SMA is rare with late-onset disease manifesting in patients aged >35 years.
The survival motor neuron (SMN) gene located on chromosome 5q is associated with SMA. There are two nearly identical genes called the telomeric SMN1 and tandemly duplicated centromeric SMN2 present at the locus. SMN2 is distinguished from SMN1 based on the difference in a single nucleotide that disrupts an exonic splice enhancer in exon 7 and results in lack of exon 7 and production of unstable (Burnett et al., 2009) and less functional proteins (Le et al., 2005; Butchbach, 2016). Over 95% of patients show deletion of SMN1. A few patients with adult-onset disease may show mutations in SMN1 instead of deletion of SMN1. Symptom severity is inversely correlated with the number of copies of SMN2 (Feldkotter, 2002) in patients who lack SMN1.
Muscle biopsies obtained from patients with SMA types I and II are histopathologically indistinguishable. Group atrophy is invariably identified; however, atrophic fibers usually appear round rather than angular (Fig. 8).
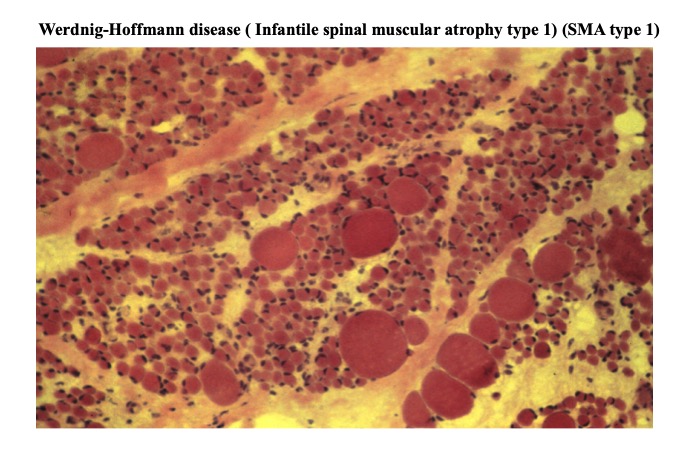
Fig.8
In neurogenic muscular atrophy, profile of atrophied fiber is not necessarily angular, as illustrated in this case of Werdnigh –Hoffmann disease (spinal muscular atrophy type I). The most important point of neurogenic change is a clear difference in diameters of atrophic and non-atrophic muscle fibers.
Although type 1 fibers predominate, this picture is often indistinguishable from fiber type grouping of a large size. Increased numbers of internal nuclei, target and split fibers, and other architectural changes are rare.
Myopathological changes in patients with SMA types III and IV show significant variations. Marked variations may be observed in different muscles in a single patient, as shown in the representative case presented in the virtual slide. Group atrophy with extensive fiber-type grouping may be observed although mild abnormalities (other than fiber-type grouping without notable atrophic fibers) may be noted in a few cases. Target fibers and other architectural changes are uncommon.




