Hereditary myopathies
Other hereditary myopathies
In this section we describe two groups of diseases that are classified based on symptomatic or histopathological features. However, it should be noted that each group includes a variety of conditions belonging to different categories of hereditary myopathies.
1. Distal myopathies
Four diseases are included under this category based on the distribution of muscle atrophy.
(1) Welander distal myopathy
Welander first described late adult-onset AD distal myopathy in Scandinavia (Welander, 1951) that typically manifests in the hands between ages 20 and 77 (mean 47 years). It is associated with the WDM gene located on chromosome 2p13. The dysferlin gene (DYSF) is also located at this locus. A previous study has reported a mutation in the RNA binding protein T-cell intracellular antigen-1 (TIA1) gene at a locus that is centromeric to DYSF (Hackman, 2013). All patients were reported to show the same point mutation and haplotype, indicating a founder effect. TIA1 is involved in resistance to stresses. Histopathological findings include myopathy with rimmed vacuoles. Ultrastructural studies have reported tubulofilamentous inclusion bodies.
(2) Miyoshi distal muscle dystrophy
Miyoshi muscle dystrophy is a dysferlinopathy with features similar to LGMD type 2B (LBMD2B) with regard to laboratory findings, histopathological findings in muscle tissue, and genetic mutations. It differs only with regard to predominant involvement of distal groups of muscles (particularly the calf muscles). Please refer to the section describing LGMD2B for more details.
(3) Distal myopathy with rimmed vacuoles (DMRV) or GNE myopathy
This condition typically affects muscles in the lower half of the legs, particularly the anterior part (shank muscles) and manifests during the second decade of life. Slow progression of the disease subsequently affects the distal muscle of the upper limbs. It is allelic with hereditary inclusion body myopathy (ARhIBM) and quadriceps sparing myopathy in Jewish families in the Middle East and shows an AR inheritance pattern associated with mutations in the UDP-N-acetylglucosamin2-epimerase /N-acetyl mannoseamin kinase (GNE) gene (Eisenberg, 2001; Kayashima, 2002). The GNE protein is involved in the synthesis of sialic acid (Nacetyl neuramic acid) in cells.
Histopathological findings include those of myopathy with rimmed vacuoles (Fig. 38). Ultrastructural studies show tubulofilamentous inclusions.
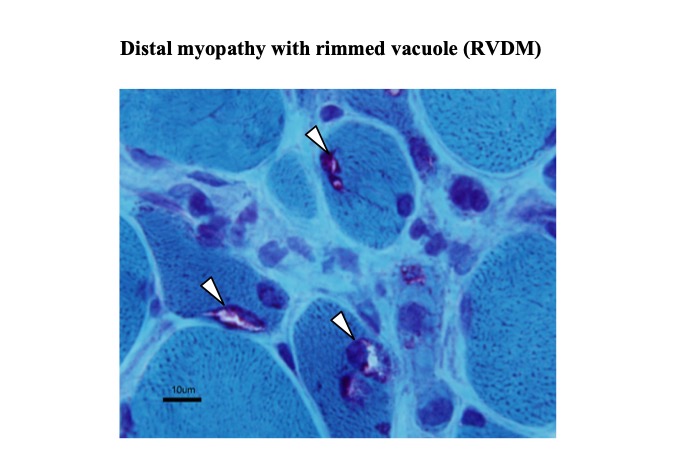
Fig.38
Distal myopathy with rimmed vacuole (arrowheads).
(4) Distal myopathy with vocal cord and pharyngeal involvement (VCPDM)
This late-onset AD distal myopathy with vocal cord and pharyngeal dysfunction was first reported as a rare disease in North America (Feit, 1998); however, this condition was also reported later from Germany (Muller, 2014) and Japan (Yamashita, 2015). It is associated with a mutation in the MATR3 gene that encodes matrin 3 and is located on chromosome 5q31 (Senderek, 2009). Matrin 3 is a component of the nuclear matrix that is expressed in skeletal muscle. Histopathological findings include rimmed vacuoles and tubular aggregates (Feit, 1998) consistent with multisystem proteinopathy.




