Hereditary myopathies
Other hereditary myopathies
2. Myofibrillar myopathies
Myofibrillar myopathies (MFM) are hereditary myopathies involving abnormalities in proteins associated with the formation and maintenance of the contractile material within muscle tissue, particularly the Z-disks, resulting in myofibrillar disorganization. The term was coined by A. G. Engel and co-workers in the 90s (Nakano, 1996; De Bleecker, 1996; Selcen, 2004). MFM is defined based on histopathological features, which include the following: (1) Trichrome-stained sections show amorphous granular or hyaline structures of various sizes and shapes, and cytoplasmic bodies and nemaline rods can be identified. Rimmed or non-rimmed vacuoles are also commonly noted. (2) Oxidative enzyme activity is reduced in areas with hyaline bodies and other abnormal structures resulting in core or rubbed-out patterns. (3) Variations are observed in fiber diameter representing myopathy. In some cases neurogenic changes may also be associated. Ultrastructural studies are characterized by a variety of abnormalities in the Z-disks, including streaming, occurrence of nemaline rods, irregular and wide expanses of areas with electron density resembling that of Z-disks. Acumulation of degraded myofibrillar material of various shapes is identified. Decreased numbers of myofibrils are commonly observed, and this change is often accompanied by an irregular increase in other organelles including mitochondria, glycogen, and lipid bodies. Mild cellular infiltration is occasionally noted.
MFM may manifest at various ages and is clinically characterized by slowly progressive muscle atrophy that is often associated with cardiomyopathy. Peripheral neuropathy may also occur in a few cases. Although AD inheritance is common, AR or XR traits are reported in some cases. MFM has been established as an important category of disease based on the myopathological viewpoint, although it is unclear where MFM is positioned in the conventional classification of muscular dystrophies and congenital myopathies.(1) Desmin-related myopathy
Desmin is an intermediate filament measuring approximately 10 nm in diameter that is present in the skeletal, cardiac, and smooth muscles. It is abundant underneath the cell surface, particularly in the vicinity of the Z-disks, in skeletal muscle. Along with other intermediate filaments, desmin structurally stabilizes myofibrils.
Clinically, desmin-related myopathy presents with chronic progressive myopathy in children and adults that is commonly associated with arrhythmias and heart failure secondary to cardiomyopathy. Distribution of muscle atrophy varies, including distal, proximal, scapuloperoneal, and facial patterns. Dysphagia and respiratory failure are sometimes noted. It presents with an AD inheritance pattern, although AR and XR inheritance is also known, and sporadic cases are common. Histopathologically, abnormal accumulation of desmin-positive material is observed in the sarcoplasm (Goebel, 1995) and deposition of granulofilamentous material is noted.(2) αB-crystallinopathy
αB-crystalline belongs to the class of heat shock proteins and is present in skeletal and cardiac muscles, as well as the lens of the eye. It forms soluble oligomers and binds to denatured proteins preventing the formation of abnormal aggregates, thereby protecting cells.
αB-crystallinopathy is typically an adult-onset AD myopathy that is often associated with cardiomyopathy and cataract. Histopathological findings are similar to those observed in cases of desmin-related myopathy including granulofilamentous inclusions (Vicart, 1998).(3) Myotilinopathy
Myotilin or myofibrillar titin-like protein (MYOT) is a Z-disk-related protein that binds to alpha actinin and filamin C. MYOT gene mutations cause LGMD type 1A (Olive, 2005). Vacuoles, filamentous inclusions, and MYOT, desmin, and alpha B crystalline aggregates are known to occur. Please also see description of LGMD1A.
(4) Zaspopathy
Z-band alternatively spliced PDZ motif-containing protein (ZASP) is a Z-disk-related protein that binds to alpha actinin. It is present in skeletal and cardiac muscles. Mutations in the ZASP gene cause myopathy with onset in patients aged >50 years. Proximal or distal atrophy may predominate in a patient and it may present with asymptomatic hyper-CKemia and may be associated with cardiomyopathy or peripheral neuropathy. Histopathological findings in muscle tissue include accumulation of desmin, αB-crystalline, gelsolin, and dystrophin. Ultrastructural findings include filamentous inclusions (Selcen, 2005).
(5) Filaminopathy
Filamin C is present in skeletal and cardiac muscles combined with Z-disk-related proteins and sarcoglycans. Filaminopathy caused by mutations in the FLNC gene is a rare adult-onset AD myopathy that primarily affects proximal groups of muscles. It may cause cardiomyopathy, peripheral neuropathy, and respiratory failure. Histopathological findings in muscle tissue include aggregates of filamin C, desmin, MYOT, Xin, dystrophin, and sarcoglycans. Ultrastructural studies may show tubulofilamentous inclusions (Kley, 2007).
(6) Bag3-opathy
The Bcl-2 associated athanogene 3 (BAG3) or CAI stressed-1 is an anti-apoptosis protein present in skeletal and cardiac muscles. It combines with HSP70 (assists with folding of several proteins) and Bcl-2, an anti-apoptotic protein and participates in signal transduction, anti-apoptosis, and protein degradation. Mutations in the BAG3 gene cause progressive myopathy in childhood associated with cardiomyopathy and respiratory failure. It may cause rigid spine syndrome and peripheral neuropathy. It shows an AD inheritance pattern. Histopathological examination of muscle specimens shows changes consistent with MFM, and electron microscopy may show myonuclei undergoing apoptosis (Selcen, 2009, 2011).
(7) FHL1-opathy
Four-and-a-half-LIM domain 1 protein (FHL 1) is a protein situated in the myofibrils and sarcolemma. Mutations in FHL 1 gene located at Xq27 causes four different muscle disorders, namely (1) reducing body myopathy, (2) X-linked myopathy characterized by postural muscle atrophy (XMPMA), (3) X-linked scapuloperoneal syndrome, and (4) Emery-Dreifuss muscular dystrophy (Schessl, 2011: Cowling,2011). It may cause rigid spine syndrome and cardiomyopathy (Schessl, 2011). These conditions are inherited as XD trait. There seems to be a great variation in severity ranging from severe infantile severe form to benign adult form. Myopathologically, reducing body (RB) is an important feature in this condition. However, RB is not seen in cases of Emery-Dreifuss muscular dystrophy and XMPMA (Cowling, 2011). Cytoplasmic bodies are often observed as a common feature of myofibrillar myopathy along with RBs.
RBs stain deep red on HE and dark purple on modified Gomori trichrome stain. They do not show NADH-TR or myosin ATPase activity, but show positive PAS reaction (Fig.39) Histochemical study for menadione-linked α -glycerophosphate dehydrogenase (MAG) activity is useful for confirmation of RB (Fig.40). Ultrastructure of RB is a cluster of randomly running filaments with small groups of glycogen granules. The clusters are often located near the myonuclei under the cell surface. No limiting membrane can be seen around the cluster (Fig.41). RBs are estimated to contain FHL1 protein (Shessl, 2009).
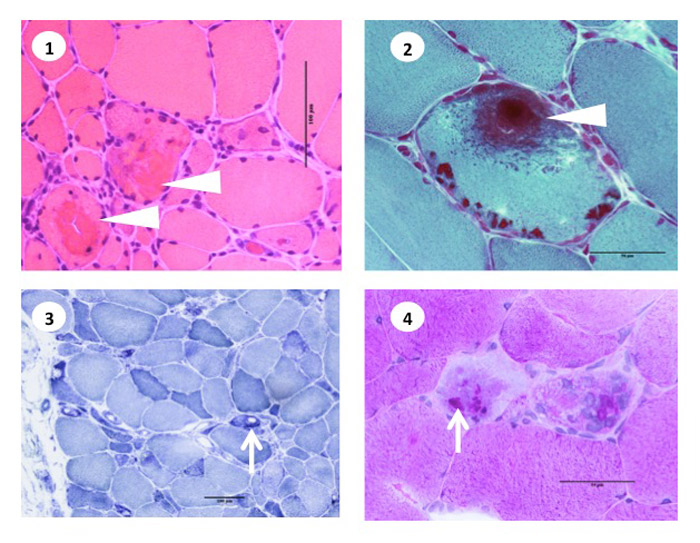
Fig.39
Recucing body myopathy
- HE:Reducing bodies (RBs) are eosinophilic cytoplasmic material (arrowhead).
- Modified Gomori trichrome: RB stains dark purple with mGT (arowhead).
- NADH-TR activity: Oxidative enzyme activity is lowered at the sites of RB (arrow).
- PAS reaction: Positive PAS rection on RBs (arrow).
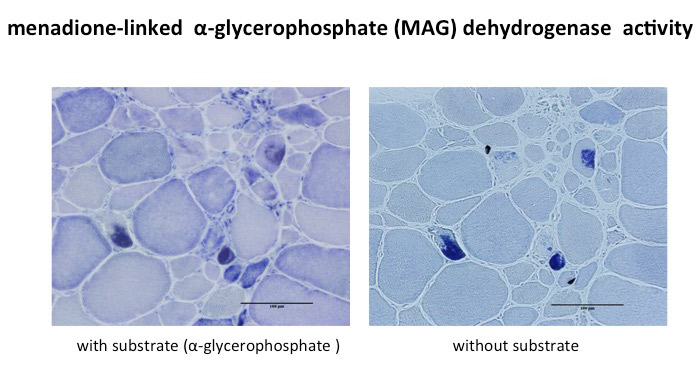
Fig.40
MAG activity in reducing body myopathy
Menadione is reduced by reducing bodies containing sulfhydryls (SH). When linked with menadione, tetrazolium is reduced to form deeply colored formazan. When combined with menadione, reducing body and tetrazolium can form formazan without substrate, α-glycerophosphate .
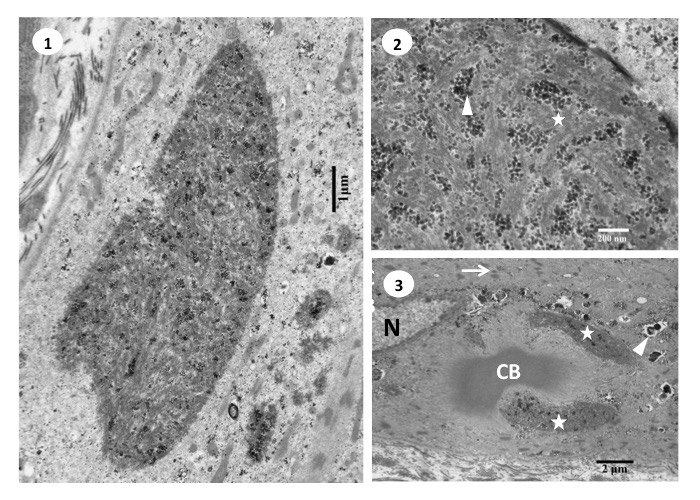
Fig.41
Ultrastructure of reducing body myopathy
- Ultrastructure of the reducing body shows large cluster of bundles of filaments running randomly with scattered groups of glycogen granules.
- Reducing body consisted of filaments (star) and glycogen granules (arrowhead). No limiting membrane is seen around the body.
- In the vicinity of a nucleus (N), a cytoplasmic body (CB) is surrounded by two reducing bodies (stars). The sarcoplasm around these structures show streaming of the Z-lines (arrow) and small rimmed vacuoles (arrowhead).
(8) Other myofibrillar myopathies
i) Myopathies with cytoplasmic bodies
Cytoplasmic bodies are a non-specific histopathological finding in muscle fibers. Ultrastructural examination indicates that they originate from Z-disk material. They occur in numerous myopathies including inflammatory myopathies, muscular dystrophies, and myofibrillar myopathies, as well as in cases of neurogenic atrophy. Cytoplasmic bodies are closely associated with the Z-disk; therefore, its distribution in the sarcoplasm often mimics that of the Z-disk. Hereditary myopathies with cytoplasmic bodies were previously referred to as cytoplasmic body myopathy, which often causes respiratory failure early in the course of illness. Such clinical features are reported in desmin-related myopathies, ACTA1-related myopathies (Donkervoort, 2017) and myopathy with early respiratory failure with mutations in the A-band domain of the TTN gene (Izumi, 2013). They show histopathological features of myofibrillar myopathy. Details of hereditary myopathy with early respiratory failure (HMERF) remain unclear.




