Hereditary myopathies
Muscular dystrophies
7. Myotonic dystrophy
Myotonia refers to delayed relaxation of skeletal muscle following contraction (Fig. 22).
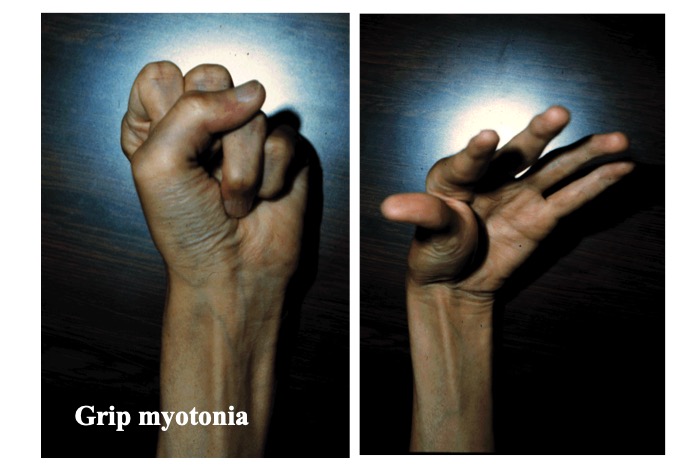
Fig.22
Grip myotonia is observed as a delayed opening of the fingers after firm grip.
It can be recorded using nEMG as prolonged repetitive myotonic discharge. Most patients with muscular dystrophy with myotonia present with type 1 myotonic dystrophy (MD), and type 2 disease that was originally described in Europe is relatively rare.
(1) MD type 1 (MD1)
MD type 1 (MD1) shows an AD inheritance pattern. Disease onset varies widely between fetal life and late adulthood. However, congenital myotonic dystrophy (CMD) with onset in utero should be distinguished from MD showing onset in childhood considering the distinctively different symptoms observed in these conditions.
CMD begins with reduced fetal movement in utero often with polyhydramnios. The infant presents with hypotonia and neonatal respiratory distress with feeding difficulties, as well as facial diplegia, a tent-shaped mouth, and arthrogryposis. Mental retardation and delayed motor development soon become evident, although myotonia may not manifest clearly. The mother invariably shows some symptoms of MD, although these are relatively mild, and it is often the patient’s grandfather who shows evidence of MD. This phenomenon may be explained by the hypothesis of larger expansion during male transmission (Brunner, 1993).
The symptoms of MD1 (childhood and adult types) tends to manifest at a younger age in the following generations in a family; this phenomenon is called “anticipation” and is attributable to elongation of CTG repeat during spermatogenesis and oogenesis. Muscle atrophy and weakness are usually conspicuous in the face, neck, and limbs. Additionally, cardiomyopathy, arrhythmia, cataract, baldness, and diabetes mellitus are commonly observed in addition to moderate degrees of mental retardation. Serum CK levels are mildly elevated. Typically, nEMG shows myotonic discharge along with myopathic changes on volitional contraction.
Abnormal extension of the CTG repeat in a non-coding region of the dystrophia myotonica protein kinase (DMPK) gene on chromosome 19q is associated with MD1. Extended CUG repeat of the transcribed pre-messenger RNA disturbs the splicing of RNA in the process of translation of several genes, specifically, the voltage-sensitive chloride channel 1 (CLCN1), the ryanodine receptor 1 (RYR1), myotubularin-related protein 1 (MTMR1), insulin receptor (INSR), troponin T2 (TNNT2), glutamate ionotropic receptor NMDA type subunit 1 (GRIN1), pre-amyloid beta A4 (APP), and the microtubuleassociated protein tau (MAPT) genes. This explains the multisystemic complications associated with MD1.
Histopathological examination of muscle specimens shows significantly increased numbers of internal nuclei, prominent changes in the internal structure of muscle fibers, such as ring and split fibers, along with type 1 fiber atrophy (Fig. 23).
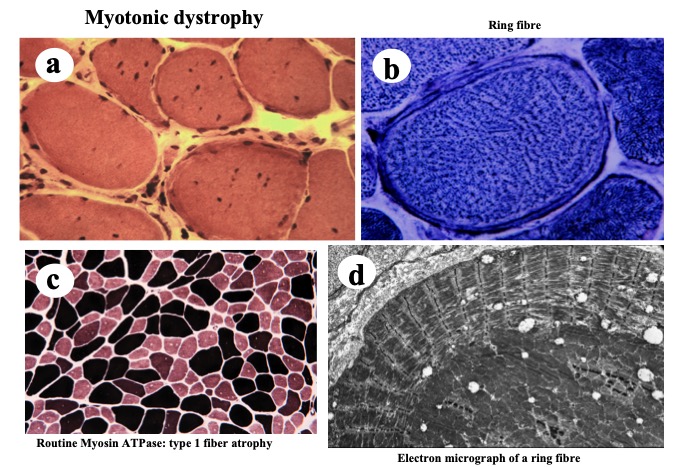
Fig.23
Myotonic dystrophy shows increase in number of internal nuclei (a), ring fibers (b: NADH-TR activity), and type 1 fiber atrophy (c: routine myosin ATPase activity). Electron microscopy demonstrates abnormal internal structure of muscle fiber corresponding to the ring fiber. Myofibrils under the cell surface are cut longitudinally whereas those in the center are cut transversely.
(2) Proximal myotonic myopathy (MD type 2) (MD2)
Compared to MD1, onset of MD2 occurs in slightly older patients and neither congenital nor childhood types of MD2 are known. It shows an AD inheritance pattern, and moderate anticipation is reported.
Proximal muscle atrophy is the main symptom; therefore, it is also referred to as proximal myotonic myopathy and may be misdiagnosed as LGMD. It is often accompanied by myalgia, and the myotonia component is usually less prominent than that observed in patients with MD1. Myotonia may be absent clinically, although it can be detected on nEMG (Ricker, 1995). Cardiomyopathy, arrhythmia, cataract, baldness, and diabetes may occur concomitantly (Ricke, 1996). So far, MD2 is rare in Japan.
Genetically, MD2 is associated with abnormal CCTG repeat expansion in the non-coding region of the ZNF6 gene (CNBP) located on chromosome 3p21. It results in abnormal expansion of CCUG repeat in the mRNA, which contributes to abnormal splicing of proteins (Raheem, 2010).
Myopathological changes are similar to those occurring in cases of MD1 except that type 2 fiber atrophy is common.




