Hereditary myopathies
Progressive hereditary myopathies are called muscular dystrophies, whereas non-progressive or slowly progressive conditions are classified as congenital myopathies. Notably, with significant variations in progression of disease observed in many conditions, the distinction between muscular dystrophies and congenital myopathies is increasingly becoming blurred.
Muscular dystrophies
1. Duchenne muscular dystrophy (X-linked recessive muscular dystrophy, Duchenne type)
(1) Pathophysiology
Duchenne muscular dystrophy (DMD) is the most frequent and severe of the dystrophinopathies, which are defined as disorders caused by mutations in the gene encoding dystrophin (DMD). Dystrophin is a cytoskeletal protein present in skeletal muscle. DMD, located on chromosome Xp21, is the largest in the human genome with 2.4 Mb of DNA containing 79 exons and large introns. Several isoforms of dystrophin are identified in cardiac muscle, cerebral cortex neurons, cerebellar Purkinje cells, the retina, and Schwann cells.
In the skeletal muscle, dystrophin is localized to the cytoplasmic face of the plasma membrane with an uneven distribution (abundant at the I-bands than at other regions within the sarcomere). The N-terminal domain of dystrophin binds to F-actin to form a complex, which links the contractile material consisted of actin and myosin to the cell surface. The C-terminal binds with the dystrophin-associated protein complex, which consists of dystroglycans and structures that support the basal lamina, forming the dystrophin axis. These structures perform the basic function of skeletal muscle to transmit the force produced by contractile material to the cell surface to induce muscle fiber contraction.
DMD is caused by mutations in the DMD gene. These mutations include deletions in approximately 66%, point mutations in approximately 33%, and duplications in a smaller percentage of patients. The mutations particularly occur at two hot spots, exon 44 and exons 2-7 and cause deletion, partial defects, dysplasia, or a decrease in the quantity of dystrophin, resulting in fragility of the cell membrane, which becomes ‘leaky” and allows influx of extracellular fluid into the muscle cell. In addition to other factors, influx of calcium ions causes abnormal contraction of the myofibrils, which is followed by muscle fiber degeneration.
(2) Symptoms and signs
DMD usually manifests in children aged 2–4 years. Frequent falls and difficulty jumping or standing from a seated position are observed, and parents may notice delayed developmental motor milestones. Weakness of proximal limb and trunk muscles are prominent. DMD is characterized by winging of the scapula (scapula alata) (Fig. 11), the Gowers’ sign (Fig. 12), lordosis of the lumbar spine, a waddling gait, pseudohypertrophy of the calves, and shortening of the Achilles tendon. Patients usually show difficulty walking after 10 years of age, and respiratory insufficiency occurs in the late teens. Variable severity of cardiac involvement is known to occur in addition to intellectual disability in a few patients.
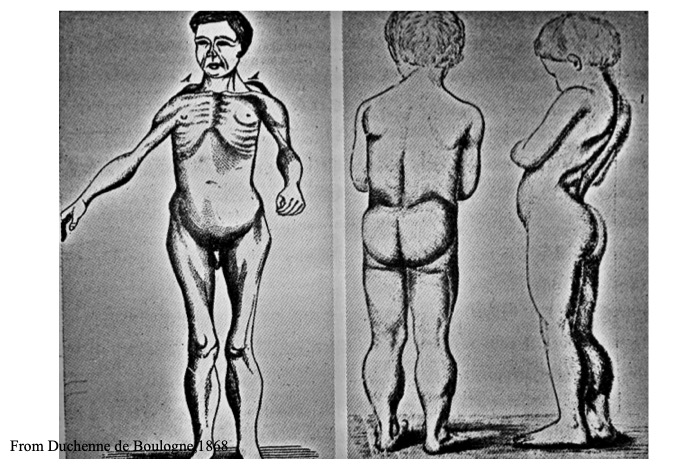
Fig.11
Duchenne’s original case, showing calf enlargement (pseudohypertrophy), winging of scapula (sacpula alata) and lumbar lordosis.
[From Arch. Cén. Méd. Vol. 11, p. 8 (1868)]
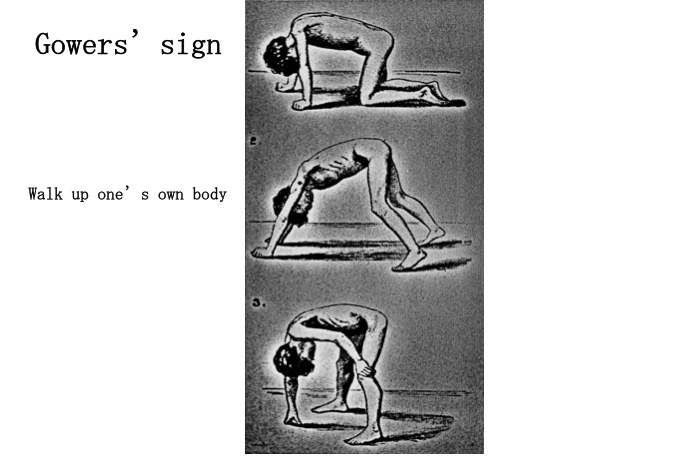
Fig.12
(3) Muscle histopathology
Typical myopathic changes include rounded muscle fibers, increased numbers of internal nuclei, and expansion of interstitial tissue. Fibrosis and fat deposition in the interstitial tissue are often observed (Fig. 13) in addition to necrotic and regenerating fibers. Opaque fibers (representing hypercontraction of the contractile material) that show intense staining with HE and modified trichrome stains are commonly identified. In the advanced stages, muscle tissue specimens show scattered islets with a collection of a small number of surviving muscle fibers interspersed amid thick connective tissue.
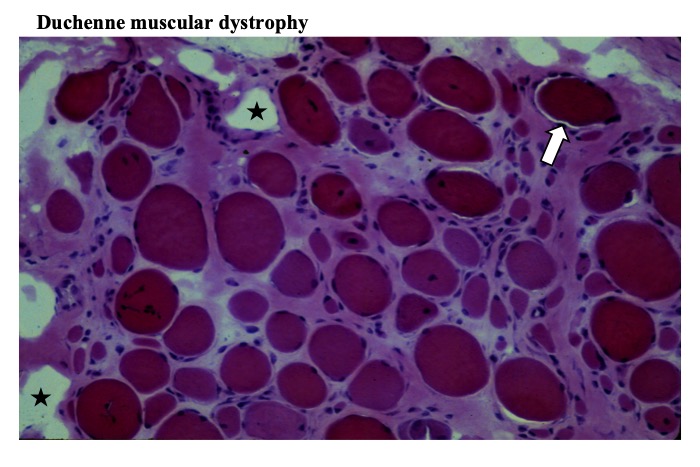
Fig.13
Muscle fibers have round contour, increased number of internal nuclei and variability in fiber diameter. Interstitial tissue is widened with fibrosis, and contain many empty spaces (stars) representing fatty deposition. Some muscle fibers called opaque fibers show dark cytoplasm (arrow) (HE stain).
Histochemical studies show non-specific architectural changes including a moth-eaten appearance and subsarcolemmal hyperactivity of mitochondrial enzymes, such as NADH-TR and SDH. Type 1 fiber predominance and atrophy are commonly noted. Type 2C fibers representing regenerating fibers are frequently noted, and identifying type 2B fibers may be difficult.
(4) Immunohistochemical analysis
Dystrophin is a large protein; therefore, its reactivity to the anti-dystrophin antibody should ideally be evaluated using two or three antibodies against different dystrophin epitopes. However, this is no longer mandatory following the availability of genetic analysis as a sensitive diagnostic tool for DMD. Normal muscle shows staining of antibodies on the surface of all muscle fibers (Fig. 14). In contrast, in muscle tissue specimens obtained from patients with DMD, there is no positive fiber (Fig. 15) excepting only a small number of muscle fibers, referred to as revertant fibers, show positive staining. The biological mechanisms contributing to the development of revertant fibers remain unclear; however, it is hypothesized that exon skipping-mediated dystrophin reading frame restoration (Lu et al., 2000) and other such mechanisms are possible contributors.
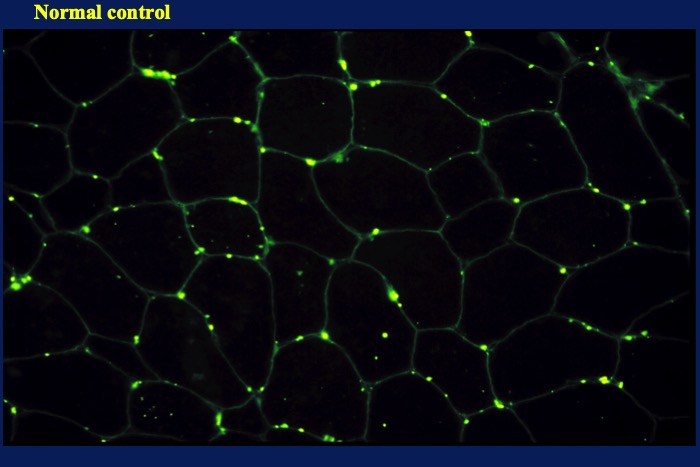
Fig.14
Immunofluorescence micrograph of normal skeletal muscle showing positivity on the surface of muscle fibers.

Fig.15
Immunofluorescence micrograph of dystrophin on a patient with Duchenne muscular dystrophy showing no positivity.
Varying degree of dystrophin expression in muscle fibers is observed in female carriers of the DMD gene (Fig. 16). Partial expression of the protein in a fiber is common. However, it is noteworthy that near-normal expression of dystrophin in all fibers may occur in most asymptomatic carriers. In contrast, it should also be remembered that non-specific and irregular weak expression of dystrophin is common in many muscular diseases. Expression of utrophin in the dystrophin-deficient portions of muscle fibers in patients with DMD may be helpful for differentiation.
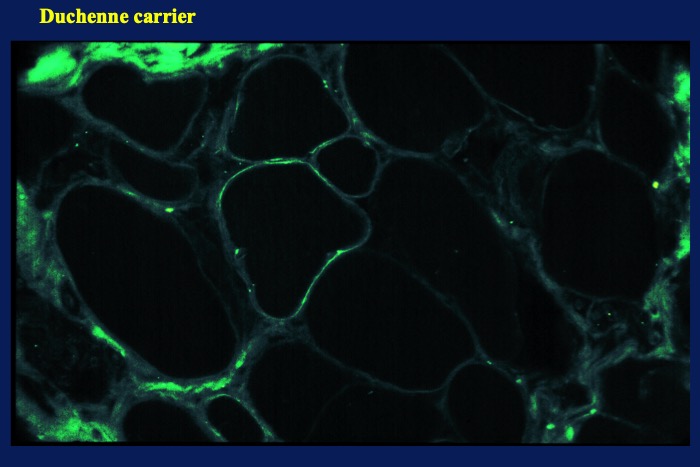
Fig.16
Dystrophin in a female carrier of Duchenne muscular dystrophy




