Hereditary myopathies
Metabolic myopathies
2. Abnormal lipid metabolism
Myopathies associated with abnormal lipid metabolism are listed in Table 3.
Disorder of lipid metabolism of skeletal muscle
- Primary carnitine deficienty
- Carnitine palmitoyltransferase deficiency (CPT)
- 2.1. CPT 1 deficiency
- 2.2. CPT 2 deficiency
- Very long chain acyl-CoA dehydrogenese (VLCAD) deficiency
- Mitochondrial trifunctional protein deficiency
- 4.1. Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency
- 4.2. MTP deficiency (combined enzyme deficiency)
- Medium–chain acyl-CoA dehydrogenase (MCAD) deficiency
- Multiple acyl-CoA dehydrogenase deficiency
- 6.1. Riboflavin-insensitive multiple acyl-CoA dehydrogenase dificiency
- 6.2. Riboflavin-responsive multiple acyl-CoA dehydrogenase deficienty
- Other primary disorders of fatty acid oxidation
- 7.1. Carnitine/acylcarnitine transolocase deficiency
- 7.2. Short-chain acyl-CoA dehydrogenase deficiency
- 7.3. Medium-chain 3-ketoacyl-CoA thiolase deficiency
- 7.4. Short-chain L-3-hydorxyacyl-CoA dehydrogenase deficiency
- 7.5. 2,4-Dienoyl-CoA reductase deficiency
- Other lipid storage myopathies
- 8.1. Muscle coenzyme Q10 deficiency
- 8.2. Chanarin-Dorfman disease or multisystem triglyceride storage disease
- 8.3. Neurtral lipid storage disease with myopathy
Tab.3
(1) Abnormalities in carnitine and related proteins
Carnitine binds to long-chain fatty acids and transports them from the cytoplasm across the outer and inner mitochondrial membranes into the matrix. This fatty acid import into cells involves the action of the carnitine transporter, carnitine palmitoyl transferase (CPT) I, CPT II, and carnitine-acylcarnitine translocase. Myopathies occur secondary to mutations in the carnitine transporter CPT II.
i) Systemic carnitine deficiency
Systemic carnitine deficiency is caused by mutations in the SLC22A2 gene, which codes organic cation/transporter 2, a protein involved in carnitine transport at the cell membrane. This condition causes severe hepatic and cardiac impairment in infants, although symptoms are milder in patients with juvenile and adult onset-disease, which shows lipid storage myopathy and cardiomyopathy. Serum CK levels are elevated in infants but not in children and adults. Serum carnitine levels are low.
ii) Carnitine palmitoyltransferase II (CPT II) deficiency
CPT II catalyzes the conversion of long-chain acylcarnitine to long-chain acyl CoA. Neonatal and infantile forms of CPT II deficiency show a severe presentation, although the myopathic type with onset in childhood or adulthood or even during middle age shows milder symptoms. The latter type is associated with intermittent episodes of myalgia and rhabdomyolysis. These episodes are induced by exercise, starvation, cold, or the administration of specific drugs. Patients are asymptomatic, and serum CK levels are normal during the interval between episodes. Histopathological examination of muscle tissue shows no abnormalities or only moderate myopathy. Measurement of serum acylcarnitine is a useful screening test; however, estimation of tissue CPT II activity is necessary for diagnosis.
(2) Abnormalities of β-oxidation
i) Very long-chain acyl-CoA dehydrogenase (VLCAD) and trifunctional protein (TFP) deficiency
Very long-chain acyl-CoA dehydrogenase (VLCAD) is located in the inner mitochondrial membrane. It initiates beta oxidation of very long-chain acyl-CoA, followed by interaction with the mitochondrial trifunctional protein (TFP), which shows three domains (long-chain 2,3-enoylCoA hydratase [LCEH], long-chain 3 hydorxy acyl CoA dehydrogenase [LCHAD], and long-chain 3-ketoacyl-CoA thiolase [LCKT]). TFP deficiency is associated with loss of function of all three domains, but in a few cases, deficiency of only LCHAD is observed.
All conditions present with severe neonatal and infantile forms causing impairment of cardiac and skeletal muscles, encephalopathy, hepatomegaly, and hypoglycemia. The adult myopathic form commonly presents with rhabdomyolysis, muscle pain and weakness. In addition, TFP deficiency can cause retinopathy and peripheral neuritis.ii) Multiple acyl-CoA dehydrogenase deficiency [MADD], (glutaric acidemia type II)
An electron transfer flavoprotein (ETF) located on the inner mitochondrial membrane plays an important role in the electron transport system by receiving electrons from dehydrogenases of the beta oxidation pathway and the branched amino acid oxidation pathway and transferring them to ETF dehydrogenase (ETFDH). Deficiency of ETF or ETFDH secondary to mutations in the ETFA, ETFB or ETFDH genes causes glutaric aciduria type II, a condition that shows an AR inheritance pattern.
Clinically, the infantile form manifests with severe hypotonia, respiratory failure and hypoglycemia, and the milder form of the disease, that shows late onset, presents with exercise-induced muscle pain or rhabdomyolysis. Histopathological examination of muscle tissue shows accumulation of lipids in both type 1 and 2 fibers with other non-specific changes. Administration of riboflavin, L-carnitine, and glycine may be beneficial for energy production in muscles.(3) Abnormalities in triacylglycerol (TG) hydrolytic enzymes
i) Lipin 1 deficiency
Lipin 1 is encoded by the LIPIN1 gene and acts as a phosphatidate phosphatase enzyme to synthesize triacylglycerol in the cytoplasm. It also acts in the nucleus as a transcriptional coactivator to control lipid metabolism and glucose homeostasis. Lipin 1 deficiency secondary to mutations in LIPIN1 causes recurrent rhabdomyolysis that is often triggered by infection and fever, rather than exercise.
Histopathological examination of muscle tissue during the interval between episodes usually shows no abnormality but may reveal fat deposition.ii) Neutral lipid storage diseases
Neutral lipid storage diseases (NLD) are caused by enzymes that catalyze the degradation of triacyl glycerol (Schweiger, 2009). Neutral lipid storage disease with myopathy (NLSDM) is caused by a mutation in the patatin-like phospholipase domain-containing protein 2 (PNPLA2) gene, which encodes adipose triglyceride lipase (ATGL) (Fischer, 2007). It causes slowly progressive myopathy occasionally associated with cardiomyopathy. Muscle biopsy shows accumulation of lipid droplets in the muscle fibers (Fig.36).
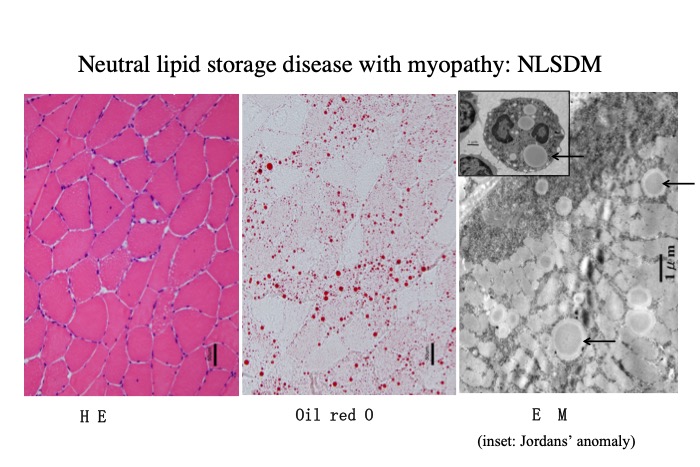
Fig.36
In NLSDM, muscle shows vacuoles of various sizes which contain accumulated lipid. Oil red O, a lipid stain, demonstrates lipid droplets stained in red. Electron microscopy shows lipid droplets (arrows) in the muscle and in the neutrophils (inset).




